The NEAT method of evolving artificial neural networks combines the usual search for appropriate network weights with complexification of the network structure. This approach is highly effective, as shown e.g. in comparison to other neuroevolution (NE) methods in the double pole balancing benchmark task . The NEAT method consists of solutions to three fundamental challenges in evolving neural network topology: (1) What kind of genetic representation would allow disparate topologies to crossover in a meaningful way? Our solution is to use historical markings to line up genes with the same origin. (2) How can topological innovation that needs a few generations to optimize be protected so that it does not disappear from the population prematurely? Our solution is to separate each innovation into a different species. (3) How can topologies be minimized throughout evolution so the most efficient solutions will be discovered? Our solution is to start from a minimal structure and add nodes and connections incrementally. In this section, we explain how each of these solutions is implemented in NEAT.
![\begin{figure}\centering
\pspic[\psline]{450pt}{!}{genome-fig_redone2-bwv2}
\t...
...nbounded.
\vspace{-\baselineskip}% Make figure a little tighter
}
\end{figure}](img3.png)
Evolving structure requires a flexible genetic encoding. In order to allow structures to complexify, their representations must be dynamic and expandable. Each genome in NEAT includes a list of connection genes, each of which refers to two node genes being connected. (Figure 2). Each connection gene specifies the in-node, the out-node, the weight of the connection, whether or not the connection gene is expressed (an enable bit), and an innovation number, which allows finding corresponding genes during crossover.
![\begin{figure}\centering
\pspic[\psline]{340pt}{!}{ij-structmutes-figure-bwv2}
...
...ulation.
\vspace{-\baselineskip}% Make figure a little tighter
}
\end{figure}](img4.png)
Mutation in NEAT can change both connection weights and network structures. Connection weights mutate as in any NE system, with each connection either perturbed or not. Structural mutations, which form the basis of complexification, occur in two ways (Figure 3). Each mutation expands the size of the genome by adding gene(s). In the add connection mutation, a single new connection gene is added connecting two previously unconnected nodes. In the add node mutation, an existing connection is split and the new node placed where the old connection used to be. The old connection is disabled and two new connections are added to the genome. The connection between the first node in the chain and the new node is given a weight of one, and the connection between the new node and the last node in the chain is given the same weight as the connection being split. Splitting the connection in this way introduces a nonlinearity (i.e. sigmoid function) where there was none before. When initialized in this way, this nonlinearity changes the function only slightly, and the new node is immediately integrated into the network. Old behaviors encoded in the preexisting network structure are not destroyed and remain qualitatively the same, while the new structure provides an opportunity to elaborate on these original behaviors.
Through mutation, the genomes in NEAT will gradually get larger. Genomes of varying sizes will result, sometimes with different connections at the same positions. Crossover must be able to recombine networks with differing topologies, which can be difficult . The next section explains how NEAT addresses this problem.
It turns out that the historical origin of each gene can be used to tell us exactly which genes match up between any individuals in the population. Two genes with the same historical origin represent the same structure (although possibly with different weights), since they were both derived from the same ancestral gene at some point in the past. Thus, all a system needs to do is to keep track of the historical origin of every gene in the system.
Tracking the historical origins requires very little computation.
Whenever a new gene appears (through structural mutation),
a global innovation number is incremented
and assigned to that gene.
The innovation numbers thus represent a
chronology of every gene in the system. As an example,
let us say the two mutations in Figure 3
occurred one after another in the system.
The new connection gene
created in the first mutation is assigned the number ![]() , and the
two new connection genes added during the new node mutation are
assigned the numbers
, and the
two new connection genes added during the new node mutation are
assigned the numbers ![]() and
and ![]() .
In the future, whenever these genomes crossover,
the offspring will inherit the same innovation
numbers on each gene.
Thus, the historical
origin of every gene in the system is known throughout evolution.
.
In the future, whenever these genomes crossover,
the offspring will inherit the same innovation
numbers on each gene.
Thus, the historical
origin of every gene in the system is known throughout evolution.
A possible problem is that the same structural innovation will receive different innovation numbers in the same generation if it occurs by chance more than once. However, by keeping a list of the innovations that occurred in the current generation, it is possible to ensure that when the same structure arises more than once through independent mutations in the same generation, each identical mutation is assigned the same innovation number. Through extensive experimentation, we established that resetting the list every generation as opposed to keeping a growing list of mutations throughout evolution is sufficient to prevent an explosion of innovation numbers.
![\begin{figure}\centering
\pspic[\psline]{330pt}{!}{matchup-fig-bwv2}
\titledcap...
...n inherited gene is disabled if it is disabled in
either parent.
}
\end{figure}](img8.png)
Through innovation numbers, the system now knows exactly which genes match up with which (Figure 4). Genes that do not match are either disjoint or excess, depending on whether they occur within or outside the range of the other parent's innovation numbers. When crossing over, the genes with the same innovation numbers are lined up and are randomly chosen for the offspring genome. Genes that do not match are inherited from the more fit parent, or if they are equally fit, from both parents randomly. Disabled genes have a 25% chance of being reenabled during crossover, allowing networks to make use of older genes once again.1
Historical markings allow NEAT to perform crossover without the need for expensive topological analysis. Genomes of different organizations and sizes stay compatible throughout evolution, and the problem of comparing different topologies is essentially avoided. This methodology allows NEAT to complexify structure while still maintaining genetic compatibility. However, it turns out that a population of varying complexities cannot maintain topological innovations on its own. Because smaller structures optimize faster than larger structures, and adding nodes and connections usually initially decreases the fitness of the network, recently augmented structures have little hope of surviving more than one generation even though the innovations they represent might be crucial towards solving the task in the long run. The solution is to protect innovation by speciating the population, as explained in the next section.
NEAT speciates the population so that individuals compete primarily within their own niches instead of with the population at large. This way, topological innovations are protected and have time to optimize their structure before they have to compete with other niches in the population. In addition, speciation prevents bloating of genomes: Species with smaller genomes survive as long as their fitness is competitive, ensuring that small networks are not replaced by larger ones unnecessarily. Protecting innovation through speciation follows the philosophy that new ideas must be given time to reach their potential before they are eliminated.
Historical markings make it possible for the system to divide
the population into species based on topological similarity.
We can measure the distance ![]() between two network encodings as
a linear combination of the number of excess (
between two network encodings as
a linear combination of the number of excess (![]() ) and disjoint
(
) and disjoint
(![]() ) genes, as well as the average weight differences of matching
genes (
) genes, as well as the average weight differences of matching
genes (![]() ):
):
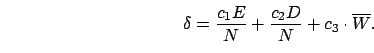
The coefficients ![]() ,
, ![]() , and
, and ![]() adjust the importance of the
three factors, and the factor
adjust the importance of the
three factors, and the factor ![]() , the
number of genes in the larger genome,
normalizes for genome size (
, the
number of genes in the larger genome,
normalizes for genome size (![]() can be set to 1 if both genomes are
small).
Genomes are
tested one at a time; if a genome's distance to a randomly chosen
member of the species is less than
can be set to 1 if both genomes are
small).
Genomes are
tested one at a time; if a genome's distance to a randomly chosen
member of the species is less than ![]() ,
a compatibility threshold,
it is placed into this species.
Each genome
is placed into the first species from the previous generation
where this condition is satisfied,
so that no genome is in more than one species. If a genome
is not compatible with any existing species, a new species is created.
The problem of choosing the best value for
,
a compatibility threshold,
it is placed into this species.
Each genome
is placed into the first species from the previous generation
where this condition is satisfied,
so that no genome is in more than one species. If a genome
is not compatible with any existing species, a new species is created.
The problem of choosing the best value for ![]() can be avoided
by making
can be avoided
by making ![]() dynamic; that is, given a target number of
species, the system can raise
dynamic; that is, given a target number of
species, the system can raise ![]() if there are too many
species, and lower
if there are too many
species, and lower ![]() if there are too few.
if there are too few.
As the reproduction
mechanism for NEAT, we use
explicit fitness sharing ,
where
organisms in the same species
must share the fitness of their niche.
Thus, a species cannot
afford to become too big even if many of its organisms perform
well. Therefore, any one species is unlikely to take
over the entire population, which is crucial for
speciated evolution to maintain topological diversity.
The adjusted fitness ![]() for organism
for organism ![]() is calculated according to its distance
is calculated according to its distance
![]() from every other organism
from every other organism ![]() in the population:
in the population:

The sharing function ![]() is set to
is set to ![]() when distance
when distance
![]() is above the threshold
is above the threshold ![]() ; otherwise,
; otherwise,
![]() is set to 1 .
Thus,
is set to 1 .
Thus, ![]() reduces to
the number of organisms in the same species as organism
reduces to
the number of organisms in the same species as organism ![]() .
This reduction is natural since species are already clustered by
compatibility using the threshold
.
This reduction is natural since species are already clustered by
compatibility using the threshold ![]() . Every species is
assigned a potentially different number of offspring
in proportion to the sum of adjusted
fitnesses
. Every species is
assigned a potentially different number of offspring
in proportion to the sum of adjusted
fitnesses ![]() of its member organisms.
Species reproduce by first eliminating
the lowest performing
members from the population. The entire population is then replaced
by the offspring of the remaining organisms in each
species.
of its member organisms.
Species reproduce by first eliminating
the lowest performing
members from the population. The entire population is then replaced
by the offspring of the remaining organisms in each
species.
The net effect of speciating the population is that structural innovation is protected. The final goal of the system, then, is to perform the search for a solution as efficiently as possible. This goal is achieved through complexification from a simple starting structure, as detailed in the next section.
.
Thus, ![]() reduces to
the number of organisms in the same species as organism
reduces to
the number of organisms in the same species as organism ![]() .
This reduction is natural since species are already clustered by
compatibility using the threshold
.
This reduction is natural since species are already clustered by
compatibility using the threshold ![]() . Every species is
assigned a potentially different number of offspring
in proportion to the sum of adjusted
fitnesses
. Every species is
assigned a potentially different number of offspring
in proportion to the sum of adjusted
fitnesses ![]() of its member organisms.
Species reproduce by first eliminating
the lowest performing
members from the population. The entire population is then replaced
by the offspring of the remaining organisms in each
species.
of its member organisms.
Species reproduce by first eliminating
the lowest performing
members from the population. The entire population is then replaced
by the offspring of the remaining organisms in each
species.
The net effect of speciating the population is that structural innovation is protected. The final goal of the system, then, is to perform the search for a solution as efficiently as possible. This goal is achieved through complexification from a simple starting structure, as detailed in the next section.
Unlike other systems that evolve network topologies and weights , NEAT begins with a uniform population of simple networks with no hidden nodes, differing only in their initial random weights. Speciation protects new innovations, allowing topological diversity to be gradually introduced over evolution. Thus, because NEAT protects innovation using speciation, it can start in this manner, minimally, and grow new structure over generations.
New structure is introduced incrementally as structural mutations occur, and only those structures survive that are found to be useful through fitness evaluations. This way, NEAT searches through a minimal number of weight dimensions, significantly reducing the number of generations necessary to find a solution, and ensuring that networks become no more complex than necessary. This gradual production of increasingly complex structures constitutes complexification. In other words, NEAT searches for the optimal topology by incrementally complexifying existing structure.
In previous work, each of the three main components of NEAT (i.e. historical markings, speciation, and starting from minimal structure) were experimentally ablated in order to demonstrate how they contribute to performance . The ablation study demonstrated that all three components are interdependent and necessary to make NEAT work. In this paper, we further show how complexification establishes the arms race in competitive coevolution. The next section describes the experimental domain in which this result will be demonstrated.