Notung 2.6 : A Manual
Dave Danicic, Dannie Durand, Aiton Goldman,
Maureen Stolzer, Benjamin Vernot
Date:

Notung offers a unified framework for incorporating duplication-loss parsimony into phylogenetic tasks. This parsimony principle asserts that gene duplication and gene loss are rare events. Notung’s functions embody the assumption that, in the absence of information from other sources, the phylogenetic hypothesis that requires the fewest duplications and losses to explain the data is preferred.
Notung can:
Notung differs from other reconciliation software in that it is the first and only software to reconcile and root non-binary gene trees with binary species trees and binary gene trees with non-binary species trees in addition to traditional analysis with binary gene trees and binary species trees. Another novel feature is Notung’s ability to rearrange and resolve non-binary gene trees.
The specific functions that Notung can perform on each combination of inputs are given in Table 1.1.
Gene Tree Species Tree Reconcile Root Rearrange Resolve Binary Binary yes yes yes N/A Non-Binary Binary yes yes yes yes Binary Non-Binary yes yes no N/A
Table 1.1: Notung’s main functions on binary and non-binary trees.
Notung provides a graphical interface for tree manipulation and visualization and offers a command line option that can be used for automated analysis of a large number of trees.
Notung utilizes novel, efficient algorithms [, , ] for reconstructing the history of gene duplications and losses, for rooting gene trees based on duplication/loss parsimony and for the rearrangement of weakly supported areas of gene trees.
More information about Notung can be found at:
More information about other Durand Lab projects can be found at:
Notung can be used to address a broad range of applications. It can assist scientists who wish to bring gene duplication models to bear on gene tree construction; evolutionary biologists studying the history of a gene family; and experimental biologists interested in incorporating evolutionary insights into questions of function and structure.
The graphical user interface was partially constructed using the tree visualization library provided by FORESTER (version 1.92) [].
NOTE: While other events besides duplication and loss, such as horizontal gene transfer, may be the cause of gene tree-species tree disagreement, Notung does not consider these other events.
D. Durand, B. V. Halldorsson, B. Vernot. A Hybrid Micro-Macroevolutionary Approach to Gene Tree Reconstruction. Journal of Computational Biology, 13(2):320-335, 2006.
B. Vernot, M. Stolzer, A. Goldman, and D. Durand. Reconciliation with non-binary species trees. Journal of Computational Biology, in press, 2008. Also appeared in Computational Systems Bioinformatics: CSB2007 Conference Proceedings, Imperial College Press: 441-452.
This manual provides a detailed description of Notung, and gives step-by-step instructions for Notung’s tasks and visualization features. It assumes familiarity with basic concepts of phylogeny reconstruction. For more information on these subjects, refer to basic textbooks, such as [, ]. A Glossary is provided. Additional sources are provided in the Bibliography.
The manual is organized into numbered chapters by topic. Each chapter begins with paragraphs describing the topic, followed by a list of step-by-step commands for operations associated with the topic. Figures showing the Notung graphical user interface (GUI) have been included to illustrate program displays and command results.
Instructions for downloading Notung for various operating systems is provided in Chapter 2. A basic introduction to the Notung GUI is provided in Chapter 3. A brief discussion regarding the relevant evolutionary theories regarding non-binary species and gene trees will be provided in Chapter 4. Notung’s six task modes are described in Chapter 5 - Chapter 10. Chapter 11 discusses the options for changing the appearance of the tree. Detailed information regarding batch processing of trees using the command line is located in Chapter 12. More detailed information about input/output and tree file formats are given in the Appendix A.
The Notung package can be downloaded from the Notung website in the file Notung-2.6.zip. When the file is unzipped, it will create a folder called Notung-2.6 that includes: this manual; a folder of sample trees which contains a folder of a sample batch run; and the Notung program file, Notung-2.6.jar.
Notung is supported on Windows 2000, Windows XP, and Windows Vista; Mac OS X 10.3 and above; and Linux. To run Notung, Java must be installed on your computer. Notung has been tested under Java 1.4.2, but should work for newer versions of Java.
To download Notung-2.6:
To unzip Notung-2.6.zip:
On Windows:
On Mac OS X:
On Linux:
unzip Notung-2.6.zip
If you do not know if you have Java:
java -version
Notung requires at least Java 1.4.
To get Java (if you do not have it):
Notung is a tool for comparing gene and species trees. Notung takes tree files as input and allows users to refine and manipulate them. The modified trees can be saved as output. The following subsections introduce basic input and output in Notung, general tree statistics, the graphical user interface, and the parameter values used in Notung’s tree refinement tasks.
To perform its functions, Notung requires a gene tree and a species tree. The species tree must contain all the species from which genes in the gene tree were sampled. The species tree may contain additional species as well - these will be ignored. A correspondence between the leaves of the species and gene trees is determined by comparing the leaf labels in the gene and species trees: each leaf label in the gene tree must include a substring that specifies the species from which the gene was sampled. Trees may be provided in Newick, NHX, or Notung format. See Appendix A - File Formats for further information.
Notung can operate on a non-binary gene tree or a non-binary species tree. However, its functions cannot be performed when both the gene tree and corresponding species tree are non-binary. For a complete summary of functions that Notung can perform, see Table 1.1.
NOTE: If you are interested in using Notung to analyze non-binary trees, see Chapter 4 - Non-Binary Trees for more a more detailed and theoretical discussion on non-binary trees.
The species tree must be rooted, with leaf nodes labeled with species names. Internal nodes may be given taxonomic labels (e.g., “tetrapoda”), but this is not required. If the internal nodes are not labeled, Notung will assign alphanumeric labels (such as n1, n2, etc.). If the species tree has edge weights or branch lengths, this information will be ignored. For more information on species names, see Appendix ?? - Specifying the Species Associated with Each Gene.
The tasks that Notung performs are based on the assumption that the user has selected a species tree that is a reliable representation of the true species relationships. Using Notung with an incorrect species tree will give incorrect results. For more information on selecting an appropriate species tree, see Chapter B - Building a Species Tree.
In order to perform its reconcile, rearrange and resolve functions, Notung requires a rooted gene tree. If the gene tree is not rooted, Notung can be used to root the gene tree. See Chapter 6 - Rooting Mode. The leaf nodes in the gene tree must be labeled with a unique identifier specifying the gene, as well as the species from which the gene was sampled. See Appendix ?? - Specifying the Species Associated with Each Gene for more information. The internal nodes may be labeled. If the internal nodes are not labeled, Notung will assign alphanumeric labels (e.g. n5, n6, etc.).
In Rearrangement mode, Notung requires that the tree have edge weights. These are used to identify edges that are weakly supported and may be rearranged. These weights may be bootstrap values, posterior probabilities, edge lengths, or any other weighting scheme selected by the user. Several different fields in the Newick and NHX formats may be used to store edge weights. See Appendix ?? - File Formats for a detailed explanation of these formats and how to indicate to Notung which field is being used for edge weights in a particular input tree.
Many tree reconstruction programs represent an unrooted binary tree as a mostly binary tree, with a single trifurcation at the root. Unless a root is selected for these trees (in Notung or another program), Notung will incorrectly treat them as rooted non-binary trees. If such a tree is actually an unrooted binary tree, failing to root it will affect Notung’s diagnostics. See Chapter 6 - Rooting Mode for more information on rooting gene trees.
Notung’s graphical interface facilitates tree visualization and manipulation, enabling the user to inspect duplicated and lost nodes in a tree, view orthologs and paralogs, visualize alternate optimal trees, and color annotate genes for visual differentiation or presentation.
To run Notung:
Using the graphical user interface on Windows or Mac OS X:
Using the graphical user interface on Linux:
java -jar Notung-2.6.jar
In addition, Notung can perform many of its operations from the command line without launching the GUI. See Chapter ?? - Command Line Options and Batch Processing for a description of the command line interface.)
When Notung is first launched, the program window will be blank. Figure 3.1a and Figure 3.1b show Notung’s graphical interface once a gene tree and species tree have been opened. Notung’s graphical user interface has the following components:
Tree panel: The tree that is currently selected appears in the tree panel. Trees are rendered with the root at left and leaf nodes at right. Nodes are denoted by small blue squares in the tree. Edge weights and leaf node names appear in the tree by default. Notung fits the whole tree in the tree panel by default. The size of the tree and tree labels can be modified using the Zoom and Fonts menus, respectively. See Chapter 11 - Changing the Appearance of the Tree Panel.
Click on image to see larger version[][]

Figure 3.1: Notung’s graphical user interface displaying (a) a gene tree, and (b) a species tree. The tree panel is highlighted in red, the task panel in blue, and the parameters panel in yellow. Only the tree panel and the task panel are applicable to species trees.
Although multiple trees can be open in Notung at once, Notung operates on only one tree at a time. To facilitate working with many trees, Notung marks each open tree with a tab at the top of the tree panel. Clicking on a tab selects the corresponding tree. Tabs are labeled with the file name and special icons to identify them as a gene or species tree - a DNA helix for gene trees, and a cartoon of the evolution of humankind for species trees (see Figure 3.2).
Click on image to see larger version
Figure 3.2: Tree tabs for a gene tree (left) and a species tree (right)
Task panel: Operations on the tree are performed in the task panel (highlighted in blue in Figure 3.1). Tabs at the top of the task panel correspond to the various tasks that Notung can perform. Clicking on a tab puts Notung in the corresponding task mode, revealing the buttons that control tasks specific to that mode. If a gene tree is selected, six modes are available: History, Reconciliation, Rooting, Rearrange, Resolve, and Annotations. Only the History and Annotation modes can be used when a species tree is selected.
Parameter values: When a gene tree is selected, a box displaying the Edge Weight Threshold and Costs/Weights for Duplications, Conditional Duplications, and Losses appears in the bottom-right corner of the program window. These values can be changed by clicking the “Edit Values” button directly below them. Note that when a species tree is selected, the program window will not display the parameter values.
Notung can read and save tree files in Newick, NHX, and Notung file formats. NHX and Notung file formats are extensions of Newick; See Appendix A - File Formats for details. Notung can also save the image in the tree panel as a Portable Network Graphic (PNG) file.
To open trees:
NOTE: Notung cannot distinguish gene trees from species trees automatically. If a gene tree is opened as a species tree, or a species tree is opened as a gene tree, reconciliation will produce incorrect results.
To save trees:
NOTE: The default format for saving trees is the Notung File Format. If you have modified the tree in Notung and wish to reopen this tree in Notung, it may be best to save the tree in Notung format. If you wish to reopen the modified tree in another tree program, Newick format may be a better option.
To view text formatted trees in a dialog box:
To copy this information, click the “Copy to clipboard” button. This text can then be pasted in any text editor.
NOTE: Selecting “About Tree Formats” from the drop-down menu will provide a dialog box containing a summary on the different tree formats. See Appendix A - File Formats for more information.
To save the current view of a tree as a PNG file:
NOTE: This option saves only the image currently visible in the tree panel. If you have zoomed in on a tree, the PNG will save only the section in view.
To save an image of the whole tree as a PNG file:
NOTE: This option saves a “pretty print” version of the entire tree. Currently, display options set in Notung will not affect the output of this tree. More options for saving tree images are available via the command line, and are discussed in Section 12.4
To print an image of a tree:
NOTE: For most printers the default page layout will be portrait; however, the landscape layout is usually preferred for printing trees from Notung. You may wish to change your printer settings before printing.
NOTE: Printing a view of the tree that shows exactly what you want may be difficult as it may be necessary to change both the printer’s settings (i.e. page layout, margins, etc.) and the appearance of the tree so that the desired print area fits within the red rectangle. See Chapter 11.2 - Zoom for more information on zooming in and out of the tree. It may be easier to obtain the desired view by first saving the tree as a PNG image, and then editing and printing that image using another program.
To reload a tree:
Note: If the tree has been modified, a dialog box will be displayed. The dialog box will offer you one of the three following options : “Save tree”; “Reload tree without saving”; “Cancel reload”.
To export color annotations to a file:
NOTE: Exported annotations can be imported into other trees, or loaded on the command line using the option --annotationfile. For more information about color annotations, see Chapter 10 - Annotations.
To import color annotations from a file:
NOTE: Annotations can be imported from previously exported annotations files. Additionally, selecting a Notung format tree which contains annotations will import annotations from that tree. Annotations can also be loaded via the command line using the option --annotationfile. For more information about color annotations, see Chapter 10 - Annotations.
To close trees:
To quit Notung:
Notung compiles information on tree characteristics, such as height, number of leaves, number of nodes, etc. Notung reports this information in the general tree statistics box under the “About This Tree” menu. The properties examined depend on whether the given tree is a gene tree or a species tree, and whether the gene tree has been reconciled or not. A description of the possible information displayed is described below.
Figure 3.3 shows an example of the tree statistics provided for a species tree.
Click on image to see larger version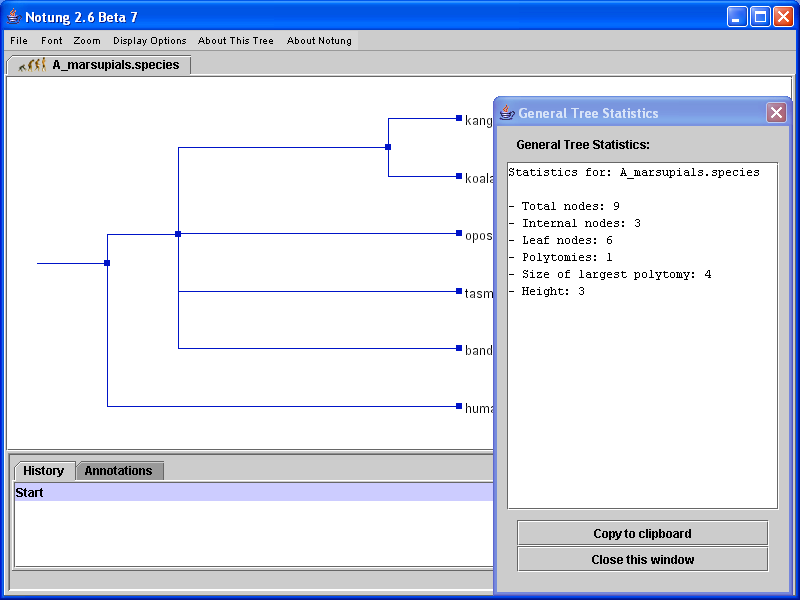
Figure 3.3: General tree statistics for a species tree.
Under the heading Reconciliation Information:
Statistics about the topology of the tree (number of leaf nodes, number of internal nodes, etc.) are reported twice: once for the gene tree without losses, and once for the tree with losses.
In addition, the species tree used for reconciliation will be reported, as well as simple statistics for the pruned species tree. Figure 3.4 shows an example of the tree statistics displayed for a reconciled gene tree.
Click on image to see larger version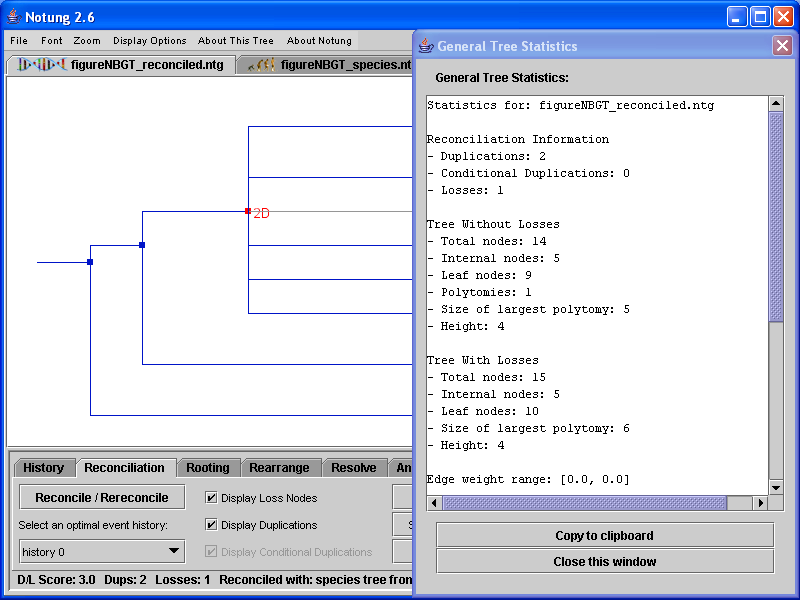
Figure 3.4: General tree statistics for a reconciled gene tree.
To get general statistics for a tree:
A window will appear containing information on the tree’s characteristics, as described above. To copy this information into your favorite text editor, click the “Copy to Clipboard” button, and paste in the text editor.
NOTE: Information on duplication bounds and losses can also be gathered through the About This Tree Menu with Duplication Bounds and Loss Counts. For more information on duplication bounds, see Chapter 12.2 - Duplication Bounds and Loss Information.
The parameter values used in Notung - the Edge Weight Threshold, Duplication Cost, Conditional Duplication Cost, and Loss Cost - can be specified by the user. These values influence the results produced by Notung’s tasks.
Notung uses a Duplication/Loss Score to score reconciled trees and evaluate alternate hypotheses. The D/L Score is defined to be: cL L + cD D + cC C where L is the number of losses, D is the number of duplications and C is the number of conditional duplications implied by the current reconciliation. The loss cost, cL, duplication cost, cD, and conditional duplication cost, cC reflect the relative importance of losses, duplications, and conditional duplications in scoring the tree. The cost of conditional duplications is only relevant when reconciling a gene tree with a non-binary species tree (see Chapter 4 - Non-Binary Trees). The default values are 1.0 for losses, 1.5 for duplications, and no cost for conditional duplications, but these values can be changed by the user. Notung displays the D/L Score of a reconciled tree, as well as the number of losses, duplications, and conditional duplications, in the bottom-left corner of the program window (see Figure 3.5).
Click on image to see larger version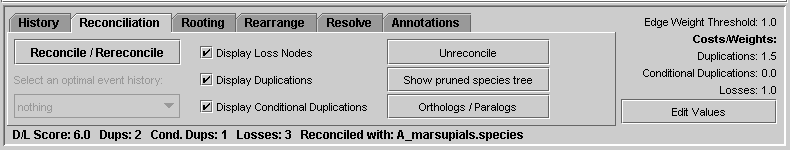
Figure 3.5: If the gene tree has been reconciled, the D/L Score, the number of duplications, conditional duplications and losses, and the species tree used to reconcile it appear at the bottom of the program window.
The Edge Weight Threshold is a parameter used to define the set of strong edges in the gene tree. In Rearrange mode, edges weighted below the Edge Weight Threshold are considered weak and may be rearranged (for more information about rearrangement, see Chapter 7 - Rearrange Mode). Edges with no weight specified are assigned an edge weight of zero, and are considered to be weak. The default threshold is 90% of the highest edge weight in the gene tree file. If no edge weights are found, the threshold is set to one. The user may change this cutoff if a different threshold is desired for the current data set.
NOTE: For some sources of edge weights, such as bootstrap values, setting the threshold to a percentage of the highest edge weight works well. For other sources, such as branch lengths, where a single very large value could cause all other edges in the tree to be weak, it may be better to set the threshold with a fixed, minimum value.
To change the parameter values:
NOTE: This will change the value settings only for the gene tree that is currently selected. Also, each history state saves the parameter values used at that state; when moving through the history, parameter values may change depending on the state and tree viewed. For more information on history states, see Chapter 9 - History.
Notung can fit a binary gene tree to a binary species tree, a binary gene tree to a non-binary species tree, or a non-binary gene tree to a binary species tree. Currently, Notung cannot compare non-binary gene trees with non-binary species trees. For a complete listing of the functions that Notung is able to perform on binary and non-binary trees, see Table 1.1.
Interpreting disagreement between gene and species trees as evidence of gene duplication and loss is widely accepted when both trees are binary. Disagreement between non-binary trees is less well-understood and there is no universally accepted approach to non-binary reconciliation. In this chapter, we briefly review current theory regarding non-binary nodes in gene and species trees and discuss how we apply these theories in Notung. If you plan to analyze either non-binary gene trees or non-binary species trees using Notung, it is recommended that you read this chapter. If you will be working solely with binary trees, you may skip ahead to the chapters describing the specific tasks you wish to perform.
For a more detailed description of Notung’s algorithm for reconciliation with non-binary species trees, see:
B. Vernot, M. Stolzer, A. Goldman, and D. Durand. Reconciliation with non-binary species trees. Journal of Computational Biology, in press, 2008.
More information on the algorithmics of reconciling, rearranging and resolving non-binary gene trees is available in:
D. Durand, B. V. Halldorsson, B. Vernot. A Hybrid Micro-Macroevolutionary Approach to Gene Tree Reconstruction. Journal of Computational Biology, 13(2):320-335, 2006.
A non-binary, or multifurcating, tree is a tree in which at least one node has more than two children. Such nodes are referred to as polytomies, or non-binary nodes. A polytomy can have several meanings []. In Notung, polytomies are represented as vertical edges with more than two children. See, for example, the polytomy in Figure 4.1.
Click on image to see larger version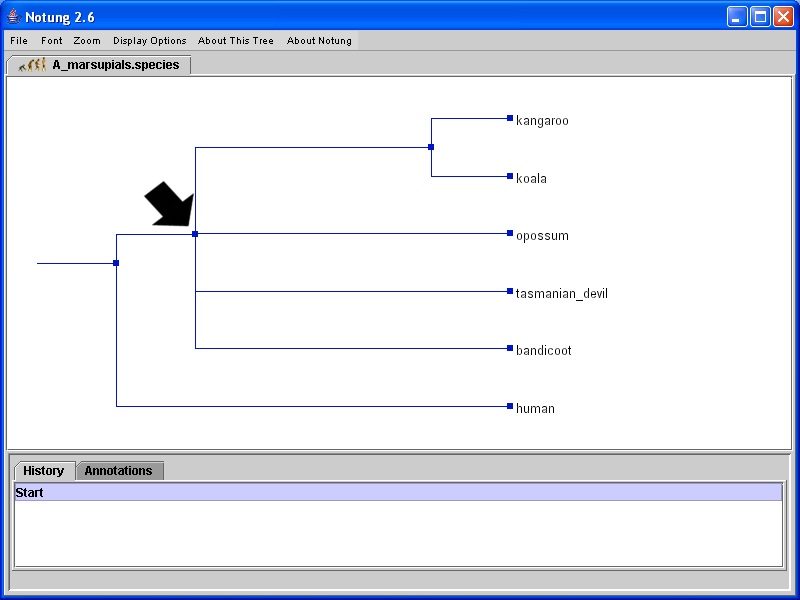
Figure 4.1: Notung displays trees as cladograms. Polytomies are drawn as vertical edges with more than two children. This tree contains only one polytomy, indicated by the arrow.
A hard polytomy represents the true, simultaneous divergence of all its children. A soft polytomy, on the other hand, refers to the situation where the true pattern of divergence is binary, but there is not enough signal in the data to determine the true branching order. Soft polytomies often occur if a sequence of binary divisions proceeds rapidly and the time between these events is insufficient to accumulate informative variation.
Reconciliation relies on the observation that discordance between a binary gene tree and a binary species tree is evidence that genes diverged through processes other than speciation. These processes include gene duplication and loss, horizontal gene transfer, and incomplete lineage sorting.
Horizontal gene transfer, the transmission of genetic material from an organism in one species to the genome of an organism in another species, is a common phenomenon in prokaryotes. The extent and importance of horizontal transfer in eukaryotes is less well-understood. Like most reconciliation software, Notung does not consider horizontal gene transfer as an explanation for disagreement when reconciling binary or non-binary trees. If you believe that horizontal gene transfer played a significant role in the data set that you plan to analyze, you may wish to consider other analysis tools.
Incomplete lineage sorting refers to discordance between gene and species trees resulting from allelic variation. Since a node in the species tree represents the evolution of a population of organisms with genetic diversity, multiple alleles may be present at the locus of interest. When lineages diverge, a different allele may fix in each lineage. The resulting gene tree will be binary and will reflect the order in which new alleles arose in the ancestral population. This pattern of divergence in the genetic lineage may not correspond to the pattern of divergence in the species lineage. For example, Figure 4.2 shows three different binary branching processes of a gene tree in the context of a species polytomy.
Click on image to see larger version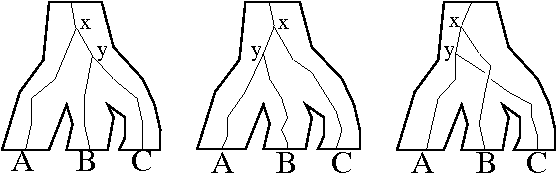
Figure 4.2: Three possible outcomes of the evolution of a single genetic locus in the context of a population. Different gene families associated with the same species polytomy may have different binary branching patterns.
A true divergence between two genetic lineages corresponds to the point where allelic differences arose, not the time of speciation. Genetic divergence that greatly predates the time of speciation is referred to as deep coalescence. In Figure 4.2, for example, the divergence at x occurs much earlier than the separation of species A, B, and C, and represents deep coalescence.
The probability of incomplete lineage sorting decreases as the time between speciation events increases [, , , , ]. If branch lengths in the species tree are sufficiently long, the effect of incomplete lineage sorting on discordance between gene and species trees is negligible, and does not need to be considered. However, when the species tree is non-binary, incomplete lineage sorting is a plausible explanation for tree disagreement.
In the next section, we discuss how Notung deals with incomplete lineage sorting when reconciling binary gene trees with non-binary species trees. In the section following this, we discuss how Notung considers the multiple, possible binary histories represented by a polytomy in a gene tree and presents the most parsimonious set of events.
Since a species tree represents the evolution of a population of organisms, a polytomy may be either hard or soft. Hard polytomies (i.e., simultaneous divergences of three or more lineages) can result from several events, such as the isolation of subpopulations within a widespread species by sudden meteorological or geological events, or from rapid expansion of the population into open territory, resulting in reproductive isolation. Soft polytomies are frequently encountered in species trees, resulting from insufficient evidence for any particular binary branching pattern. Non-binary species trees may be common; for example, 64% of branch points in the NCBI Taxonomy Database [] have three or more children.
Notung assumes that the probability of incomplete lineage sorting is negligible when a node in the species tree is binary. In this case, disagreement between the trees is interpreted as evidence for gene duplication or loss. In contrast, incongruence between a binary node in a gene tree and a non-binary node in a species tree can be evidence of either deep coalescence or gene duplication.
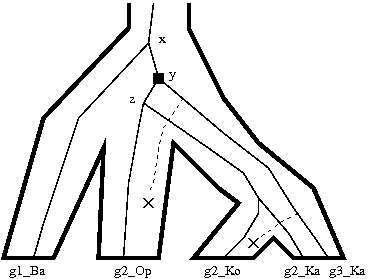
When the species tree is non-binary, Notung considers two different scenarios: cases in which disagreement can only be explained by a gene duplication (required duplications) and cases in which it is not possible to determine whether the disagreement is due to deep coalescence or gene duplication (conditional duplications). Both of these cases are illustrated in Figure 4.3.
Click on image to see larger version[][]
[]
[][]
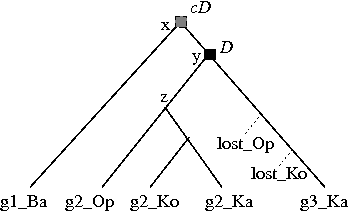
Figure 4.3: Black squares with a “D” indicate (required) duplications. Losses are represented by dotted lines. (a) A marsupial species tree with a polytomy. (b) The phylogeny of a hypothetical gene family sampled from the same marsupial species. (c) Hypothesis 1: the disagreement between (a) and (b) can be explained by deep coalescence (node x), followed by gene duplication (node y). (d) Hypothesis 2: the disagreement between (a) and (b) can also be explained by duplication at x, followed by gene loss, followed by duplication at y. (e) The divergence at x is designated a conditional duplication (gray square) because it is not possible to determine whether the disagreement is due to duplication or deep coalescence. The divergence at y is a required duplication.
Notung implements a novel reconciliation algorithm [] for non-binary species trees that distinguishes between required and conditional duplications and reports them separately.
Inferring loss events is also fundamentally different when the species tree is non-binary. When both trees are binary, an inferred loss node can always be unambiguously assigned to a specific edge in the gene tree, indicating when in the history of the gene family the loss occurred. The node is labeled with the species in which the loss occurred. However, when a loss is associated with a polytomy in the species tree, it is not generally possible to assign the loss to a single edge in the gene tree. Rather, the loss can be associated with a set of candidate edges, each of which corresponds to an alternate hypothesis regarding when the loss occurred. The inferred loss must have occurred on one of the edges in this set, but it is not possible to determine which one. Figure 4.4 shows an example of this ambiguity when assigning a gene loss in species A. This loss could be associated with any of the three colored edges indicated in Figure ??b. The three hypotheses resulting from the three possible ways of assigning the loss to an edge can be seen in Figure ??.
Click on image to see larger version[][]
[]

Figure 4.4: Losses associated with a polytomy in the species tree are ambiguous. (a) A species tree with a polytomy. (b) A gene tree drawn from the species in (a), with a loss in species A. (c) This loss can be assigned to three possible edges. Associating a loss with the green edge implies that g_A diverged first and was then lost. the blue edge implies that g_A was lost after the divergence of g_C; the red edge implies that A was lost after g_B diverged.
In a complex reconciliation with several losses, there may be many alternative hypotheses (i.e., reconciliations with different loss histories) to consider. Notung uses duplication-loss parsimony to reduce the number of candidate reconciliations. Specifically, Notung assigns each loss to a specific edge within the set of candidates, with a goal to minimize the total number of losses.
This total number of losses depends on two factors. The first is the position of the loss relative to duplications in the gene tree. Assigning a loss to an edge above a duplication implies that the loss occurred before the duplication, and only one loss is inferred. However, assigning the loss to an edge below the duplication implies that the duplication occurred first. Thus, two losses are inferred – one for each duplicated copy. Second, in some circumstances, losses in sibling species can be more parsimoniously explained by a loss in their common ancestor. The total number of losses may be reduced by assigning losses in such a way to maximize the number of cases where multiple losses can be replaced by a single loss in an ancestral species. These two factors are not independent of one another. Assigning a loss below a duplication will usually increase the total number of losses. However, in some cases, these “duplicated” losses may be combined with other losses assigned to edges below that duplication, thus reducing the total number of losses.
Two algorithms for inferring losses, one exact and the other a heuristic, have been implemented in Notung. Both algorithms are integrated with the algorithm to identify required and conditional duplications. The exact algorithm infers a history with the fewest losses, taking both of the above considerations into account. This algorithm is computationally intensive because all possible combinations of loss assignments must be considered. Its worst case running time is an exponential function of the size of the largest polytomy in the pruned species tree. In practice, the exact algorithm performs efficiently on non-binary species trees with small polytomies. However, users should be prepared for extended running times if the species tree has a polytomy with more than 12 children.
The heuristic runs significantly faster than the exact algorithm and yields the same results in many, if not most, cases. It returns only one reconciliation, which is not guaranteed to be optimal. However, in a comparison of the two methods on the 1,174 trees from TreeFam, the heuristic found an optimal solution for more than 99% of the trees. Of the seven trees where the heuristic did not find an optimal solution, in the worst case, the number of losses was overestimated by four losses from a total of 249.
NOTE: While the exact algorithm is guaranteed to return a reconciliation with a minimum number of losses, there may be more than one such optimal reconciliation; if so, Notung reports only one.
The interactive version of Notung uses the heuristic to reconcile binary gene trees with non-binary species trees. Both algorithms are available in the command line version. See Chapter ?? - Command Line Options and Batch Processing for information about these options.
In a gene tree, each lineage represents a single gene and the result of any divergence is exactly two descendant sequences. Thus, in contrast to species trees, the true branching pattern in a gene tree is always binary [], and all multifurcations are soft polytomies. For this reason, non-binary gene trees are also referred to as unresolved trees. Some phylogeny reconstruction programs output non-binary gene trees when the true binary branching process cannot be resolved. Such uncertainty often arises if binary divisions occur too rapidly to accumulate informative variation or if the data set is noisy.
Notung’s approach to reconciling non-binary gene trees rests on the assumption that the children of a polytomy arose through an unknown series of binary divergences. Notung further assumes that, in the absence of other information, the best hypothesis for the true evolutionary history of the children of the polytomy is the binary branching pattern that entails the fewest duplications and losses; there may be more than one such binary resolution of a polytomy. The problem of reconciling non-binary gene trees reduces to finding a binary tree that agrees with the original tree everywhere except at the polytomies and has a minimal D/L Score.
The general approach is as follows: A non-binary gene tree is converted into a binary gene tree by replacing each polytomy with a temporary binary resolution. This resolution is optimal under duplication-loss parsimony, when reconciled with the appropriate binary species tree. The resolution is determined by using our rearrangement algorithm [], which constructs an optimal duplication-loss parsimony tree in polynomial time per tree. Following rearrangement, all nodes and edges not present in the original gene tree are then removed, to obtain a reconciliation of the original non-binary gene tree. As nodes and edges are removed, any duplications or losses assigned to them are reassigned to their associated polytomy.
This process is illustrated in Figure 4.5. The optimal resolution of the polytomy at node z in the gene tree in (b) with the species tree in (a) is shown in the right subtree of (c). This entails one duplication and one loss. This information is mapped onto the original gene tree (b) to obtain the reconciled, non-binary gene tree in (d). The polytomy in the original tree represents uncertainty, as reflected in the reconciliation. The reconciled polytomy in the right subtree of (d) tells us that at least one duplication and one loss occurred in the subtree rooted at z, but the exact order of these events is unknown.
Click on image to see larger version[][]
[]
[]
[]

Figure 4.5: (a) A binary species tree. (b) A non-binary gene tree with genes sampled from (a). (c) Binary resolution of gene tree (b), yielding a binary tree with three duplications and three losses. (d) Gene tree (b) reconciled with species tree (a), yielding a non-binary tree with three duplications and four losses. (e) Gene tree (b) following rearrangement. The polytomy has been resolved and the weak edge has been rearranged to eliminate a duplication.
Note that multiple duplications can be assigned to a polytomy in a reconciled non-binary tree. If duplications are inferred on two or more temporary nodes in the optimal binary resolution of a polytomy, the polytomy will be assigned multiple duplications when these nodes are removed from the tree. For example, two duplications are assigned to the polytomy in the reconciled, non-binary gene tree in Figure 4.6. This differs from standard reconciliation, where every node has at most one duplication.
Click on image to see larger version[][]
[]
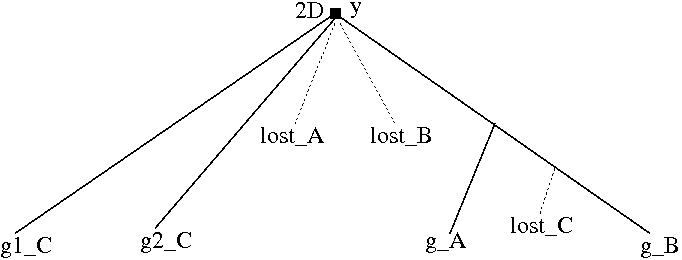
Figure 4.6: (a) A non-binary gene tree. (b) An optimal, binary resolution of gene tree (a) reconciled with species tree in Figure 4.5. (c) The reconciled non-binary, gene tree. The resulting tree has a polytomy with two duplications.
Notung can be used to infer the root of an unrooted tree by identifying the root that requires the fewest duplications and losses. In Rooting mode, when the tree is binary, each edge is assigned a root score; i.e., the D/L Score of the tree when rooted on that edge. When the gene tree is non-binary, it is also possible to root the tree on a polytomy, as shown in Figure 4.7. Placing a polytomy at the root of the tree implies that one of the edges in the true binary resolution of the polytomy is the true root.
Click on image to see larger version[][]
[]
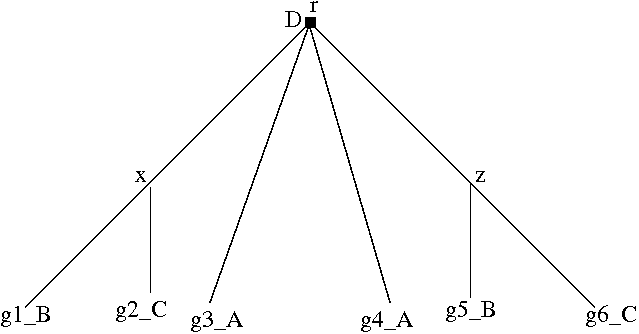
Figure 4.7: (a) An unrooted, non-binary gene tree. (b) The rooted, binary resolution of (a) with the lowest D/L Score. Rooting the tree on any other edge would entail more duplications and losses. (c) When reconciled with species tree in Figure ??, the polytomy in (a) is the root with minimum cost.
To calculate root scores, Notung roots the tree on each edge and polytomy in turn. For each root, the rearrangement algorithm is applied to ensure that each polytomy is replaced by an optimal binary resolution. The D/L Score of the resulting tree is used as the root score for that rooting. Note that it is necessary to optimize the binary resolutions separately for each root because the D/L Score depends on the location of the root. After all edges and polytomies have been scored, the original tree is reported to the user with edges and polytomies annotated with root scores.
Note that in Reconciliation and Rooting modes, binary resolutions are used to infer duplications and losses, but the structure of the final, output tree is unchanged. In the Rearrangement and Resolve modes, Notung uses duplication-loss parsimony to transform the non-binary input tree into a binary gene tree. Resolve mode is analogous to the reconciliation method described here, with the exception that the final step of removing the added nodes and edges is not performed. The result is a reconciled binary tree that is optimal with respect to duplication-loss parsimony. For the example in Figure 4.5, the Resolve function would return the tree in (c). As there may be more than one optimal resolution, Notung presents the different histories that result in the optimal tree. See Chapter 8 - Resolve Mode for more information.
In Rearrangement mode, the rearrangement algorithm is applied not only to edges added to the tree in the resolution of polytomies, but to all edges with an edge weight below the edge weight threshold. The result is a reconciled, binary tree in which weak edges have been rearranged to minimize the D/L Score. Figure 4.5(e) shows the rearrangement of the non-binary gene tree in (b), assuming an edge weight threshold of 90.
In Reconciliation mode, Notung compares a gene tree with a species tree to infer gene duplications and losses. Notung will display a reconciled tree in the tree panel with the inferred duplications and losses indicated on the tree. The D/L Score of a reconciled tree will be displayed in the lower left corner of the screen (see Figure 5.1(b)).
Click on image to see larger version[Unreconciled gene tree][Reconciled gene tree]

Figure 5.1: A binary gene tree before and after reconciliation with the species tree in Figure 3.1b.
Notung requires that gene and species trees have compatible labels, so that the species from which each gene originated can be identified. An error message will appear if one or more gene labels cannot be matched to a label in the species tree. See Appendix A.4 - Specifying the Species Associated with Each Gene for further information on gene labels.
All species represented in the gene tree must be present in the species tree, but the species tree may include additional species. During reconciliation, Notung automatically identifies the species in the species tree that are not present in the gene tree, and generates a pruned species tree with those species removed. The pruned species tree is stored in Notung’s internal data structures. This tree is not shown or saved unless the user does so explicitly.
Once a gene tree has been reconciled, Notung can infer orthologous and paralogous relationships, described in Section 5.3. Notung can also determine lower and upper bounds on the time of each duplication and conditional duplication, where bounds are represented in terms of internal nodes in the species tree; i.e., relative to speciation events. The upper bound on the time of duplication is the most recent species in which the duplication was not present. The lower bound is the oldest species in which the duplication must have been present. This information, along with statistics on losses, can be viewed in a pop-up window by selecting “Duplication Bounds and Loss Counts” from the “About This Tree” menu. Duplications and bounds in this window are identified by internal node names. For losses, each node in the species tree is listed, followed by the number of losses associated with that taxon.
Notung can reconcile binary gene trees with non-binary species trees, as well as non-binary gene trees with binary species trees. The differences between these functions and traditional reconciliation of binary gene trees with binary species trees are summarized briefly here. For a more detailed discussion of reconciliation with non-binary trees, see Chapter 4 - Non-Binary Trees. Note that orthologs and paralogs can only be inferred on binary gene trees reconciled with binary species trees.
Reconciling a binary gene tree with a non-binary species tree results in a binary gene tree with duplications and losses added. Notung distinguishes between cases in which disagreement can only be explained by a gene duplication (required duplications) and cases in which it is not possible to determine whether the disagreement is due to deep coalescence or gene duplication (conditional duplications). When reconciling a gene tree with a non-binary species tree, duplications appear in the tree as small red squares with red D’s, while conditional duplications are small pink squares with pink cD’s (see Figure 5.2).
Click on image to see larger version[Polytomy losses labeled with the names of the species from which they are absent.][Polytomy losses labeled with the number of species from which they are absent.]
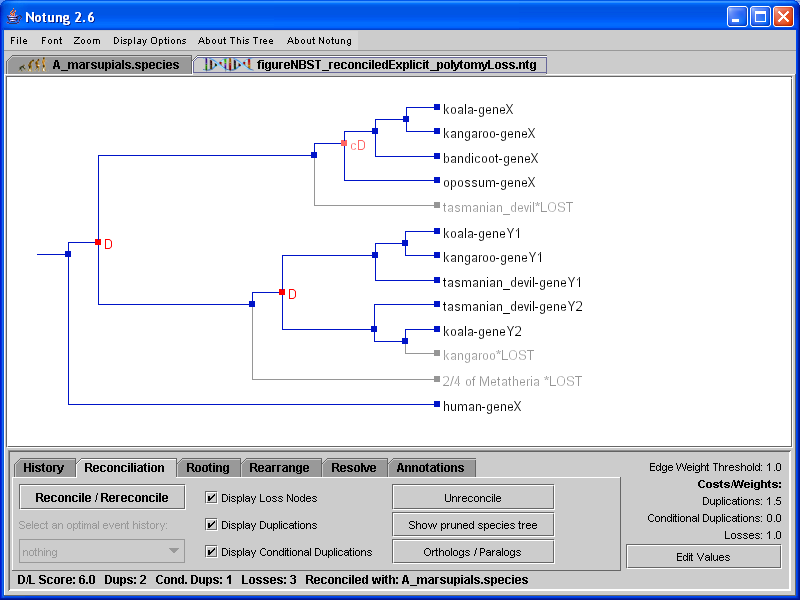
Figure 5.2: A binary gene tree reconciled with the non-binary species tree in Figure 4.1. Conditional duplications are marked by pink cD’s, while required duplications are indicated with red D’s. Polytomy losses are labeled with the name of the associated polytomy, as well as the information about the species from which they are absent.
If two or more orthologous genes are missing from species that are children of the same polytomy, then it is more parsimonious to infer a loss of the common ancestor of those genes. We refer to such losses as polytomy losses. For example, in Figure 5.2, members of the hypothetical Y gene family are missing from two species, bandicoot and opossum. These species are children of the same polytomy in the species tree in Figure 4.1. Notung infers a single loss, labeled with the names of species from which the gene is absent, as well as the label of the corresponding polytomy in the species tree. By default, polytomy losses are labeled with the species that lack the gene. However, if a polytomy loss is associated with many sibling species, the default display can produce very long labels. Users can instead opt to label polytomy losses with the number of species in which the loss occurred, as well as the label and the total number of children of the polytomy, illustrated in Figure 5.2(b).
Reconciling a non-binary gene tree with a binary species tree results in a non-binary, reconciled gene tree. A reconciled, binary gene tree can be obtained by using the Resolve function (see Chapter 8 - Resolve Mode).
Reconciliation of a non-binary gene tree with a binary species tree differs from binary reconciliation in two important ways. First, a polytomy in a non-binary gene tree may be annotated with more than one duplication. For example, the reconciled non-binary gene tree in Figure 5.3(a) has a polytomy annotated with two duplications and a loss.
Click on image to see larger version[Reconciled gene tree][Binary species tree used for reconciliation]
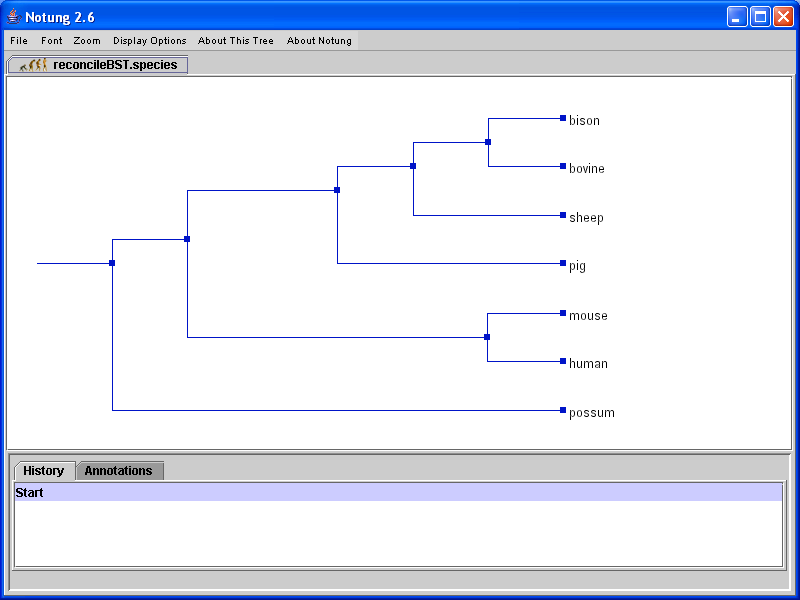
Figure 5.3: Reconciliation of a non-binary gene tree with the binary species tree in (b). More than one duplication may be inferred at polytomies in the gene tree. In addition, it is possible to have more than one optimal event history, as seen in the lower left-hand corner of the reconciliation panel in (a).
Recall that a gene tree polytomy is an indication that although its children evolved by successive binary divergences, the order in which the taxa diverged is unknown. Since this binary branching pattern is unknown, the relative order of duplications and losses with respect to those divergences cannot not be determined, either. The polytomy in Figure 5.3(a) communicates that at least two duplications and one loss occurred in the subtree descending from the polytomy, but the exact timing of those events is unknown. See Chapter 4 - Non-Binary Trees for a detailed explanation of duplications and losses in reconciled non-binary gene trees.
Second, there may be several alternate hypotheses for the reconciliation of a non-binary gene tree. Since the true binary branching pattern of a polytomy is unknown, Notung infers duplications and losses for all binary resolutions with minimal D/L Score. If there is more than one optimal binary resolution, multiple reconciliations will result. Notung addresses this issue by presenting all alternate event histories to the user. Each event history represents a different combination of duplications and losses that could result in the same minimal D/L Score. Initially, Notung arbitrarily selects one event history to present in the tree panel. The other optimal histories may be viewed using the drop-down menu labeled “Select an optimal event history,” as shown in Figure 5.3. This menu gives a list of up to 50 optimal event histories. If there are more than 50 optimal event histories, they can be generated using the Command Line Interface (see Chapter 12 - Command Line Options and Batch Processing). For a more detailed discussion of alternate event histories, see Chapter 7 - Rearrange Mode.
To reconcile a gene tree with a species tree:
If the convention selected by Notung is not the naming convention used in the gene tree, change it by selecting the appropriate radio button. See Appendix A.4 - Specifying the Species Associated with Each Gene for details about species tag specifications.
NOTE: The Prefix and Postfix formats require species names to be embedded in the gene names. NHX Species Tag format embeds the species information in a Newick comment field. When this format is used, the information will not appear on the screen unless the “Display Leaf Node Species Names” option in the Display Options menu is selected (See Chapter 11.1 - Display Options).
The reconciled tree appears in the tree panel (see Figure 5.1(b)). Duplication nodes are indicated by a square and the letter “D”, shown in red. In non-binary gene trees, the number of duplications associated with a polytomy will also be shown with a red D (e.g., Figure 5.3(a)). Loss nodes appear in light gray type and state in which species the loss occurred. A message at the bottom of the program window reminds you which species tree was used in reconciliation (e.g., “Reconciled with: <speciestreeName>”; see Figure 5.2).
To hide loss nodes/duplications:
The duplication marks or loss nodes can be hidden to avoid a cluttered image.
NOTE: When you uncheck “Display loss nodes,” Notung will reset the image so that the whole tree fits in the tree panel.
Options that are not currently available are displayed in gray type to indicate that they are disabled. In particular, the above options will be grayed out if no reconciliation has been performed. The “Display Conditional Duplications” option will also be displayed in gray if the gene tree was reconciled with a binary species tree.
To view alternate optimal event histories:
If the gene tree is non-binary, there may be more than one reconciliation. If more than one optimal event history exists for a rearranged tree, the drop down menu, “Select an optimal event history,” will be enabled.
The tree panel will now show a new tree corresponding to the selected alternate history.
If there is only one optimal history or if the tree has not been reconciled, the drop down menu will be grayed out. Recall that in Reconciliation mode multiple optimal histories are only possible when the gene tree is non-binary.
To undo the reconciliation:
To display a pruned species tree:
This option is grayed out if the gene tree has not been reconciled.
To show time bounds and information on losses:
The D/L Score of the reconciled tree appears at the top of the window, followed by duplication bounds described in three columns. The left column gives the internal node in the gene tree where the duplication occurred. The center column and right column give lower and upper bounds, respectively, on the time of duplication, expressed as node names in the pruned species tree. The total number of duplications appears below this table.
If the species tree is non-binary, conditional duplication bounds, if any, are described in the three columns below duplication bounds. The left column gives the internal node in the gene tree where the conditional duplication occurred. The center and right columns provide the lower and upper bounds, respectively, on the species tree node in which the event (duplication or allelic divergence) may have occurred. The total number of condition duplications is listed below this table.
Information on losses will appear in the two columns below the (conditional) duplication bounds. The left column lists all the nodes in the species tree. The right column gives the number of inferred losses that occurred in that species. Polytomy losses are assigned to the corresponding polytomy, rather than the individual species which lack the gene. For example, the polytomy loss in Figure 5.2 is reported as a single loss in Metatheria.
To display internal node names in the tree panel, “Display Internal Node Names” and “Display Internal Node Species Names” must be turned on in the “Display Options” menu (See Chapter 11.1 - Display Options) for both the gene and species tree.
This option is grayed out if the gene tree has not been reconciled.
To display the number of species in polytomy losses:
By default, polytomy losses are labeled with the names of the species from which they are absent.
This causes polytomy losses to be labeled with the number of children of the polytomy lost, the total number of children of the polytomy, and the name of the polytomy in which these losses occurred.
Notung can infer orthologous and paralogous relationships between genes in binary gene trees reconciled with binary species trees. Recall that two genes are orthologous if they diverged from a common ancestor via speciation. If they diverged by duplication, they are paralogous [, ]. Notung infers orthology by finding the least common ancestor of two genes in a gene tree. If that least common ancestor is a duplication node, then the two genes are paralogous. Otherwise, the two genes are orthologous.
Notung will output a matrix of pairwise orthologous and paralogous relationships in several table formats. In addition, the Notung GUI includes an interactive Ortholog/Paralog feature in the Reconciliation task panel, that allows the user to investigate these features through a point and click interface.
Orthologs and paralogs can be reported in comma-separated (CSV), tab
separated, or HTML formatted tables. For each of these options, genes
in the gene tree are listed in both column and row headers.
Orthologous genes are indicated by an “O” in the table, while
paralogous genes are indicated by a “P.” An example table, showing
orthologs and paralogs from genetree_SMALL, is shown
in Table 5.1. In HTML tables, CSS is used to color cells
representing orthologs with a blue background, and cells representing
paralogs with a pink background.
Homolog Table for: genetree_SMALL
P == Paralogous
O == Orthologous
. == Genes on X and Y axis are the same.
gB_human gA_human gA_mouse g_gorilla gB_mouse gY_cow gX_cow gB_human . P P P P O O gA_human P . P P P O O gA_mouse P P . O P O O g_gorilla P P O . P O O gB_mouse P P P P . O O gY_cow O O O O O . P gX_cow O O O O O P .
Table 5.1: An example Ortholog/Paralog table, showing orthologs and paralogs from genetree_SMALL, reconciled with speciestree_SMALL. Orthologous genes are labeld with ’O’, Paralogous genes are labeled with ’P’. Notice that this table is symmetric. Cells at the intersection of the column and row representing the same gene are labeled with ’.’.
To view an Ortholog/Paralog table:
NOTE: The selected table will be displayed in a popup dialog box. To copy the table, click “Copy to clipboard”. Tab delimited tables can usually be pasted directly into spreadsheet applications like Excel. CSV formatted tables can be opened by most spreadsheet programs via the file menu. HTML format tables can be pasted directly into web pages.
To enter the interactive Ortholog/Paralog mode, click on the “Orthologs/Paralogs” button in the Reconciliation task panel. A legend will appear in the tree panel. Mousing over or clicking on a gene will highlight it in light blue. Orthologs of this gene are highlighted in darker blue, and paralogs are highlighted in pink. The legend can be minimized by clicking on “hide”, in the legend. Click on the minimized legend to show the full legend again. The legend can be dismissed entirely by clicking “close”. The next time you enter Ortholog/Paralog mode, the legend will be visible again.
NOTE: If you use “File → Save Current View as Image (PNG)”, the image will contain the Ortholog/Paralog legend, and if a gene is currently selected, orthologs and paralogs of that gene. Currently, “File → Save Whole Tree as Image (PNG)” will not show orthologs and paralogs.
In Rooting mode, the D/L Score can be used to infer the root of a gene tree. Notung’s Rooting Analysis calculates a root score for each edge in the tree, corresponding to the D/L Score of the tree if rooted on that edge. Note that the Rooting Analysis computes root scores, but does not change the tree. The user must root the tree explicitly by clicking in the tree panel. Rooting mode can also be used to root a tree manually by clicking on any edge at any time, even if the Rooting Analysis has not been performed.
When the Rooting Analysis is complete, edges with the minimum root score are highlighted in red. Notung also highlights edges with near optimal scores in pink. Edges with scores that are greater than the minimum by at most 5 percent of the difference between the maximum and minimum score are highlighted in pink. Figure 6.1(a) shows the gene tree from Figure 5.1 after the Rooting Analysis has been applied. Note that optimal rooting edges are highlighted in red, but the gene tree topology is unchanged from Figure 5.1. Figure 6.1(b) shows the tree after it has been rooted by clicking in the tree panel.
Click on image to see larger version[][]
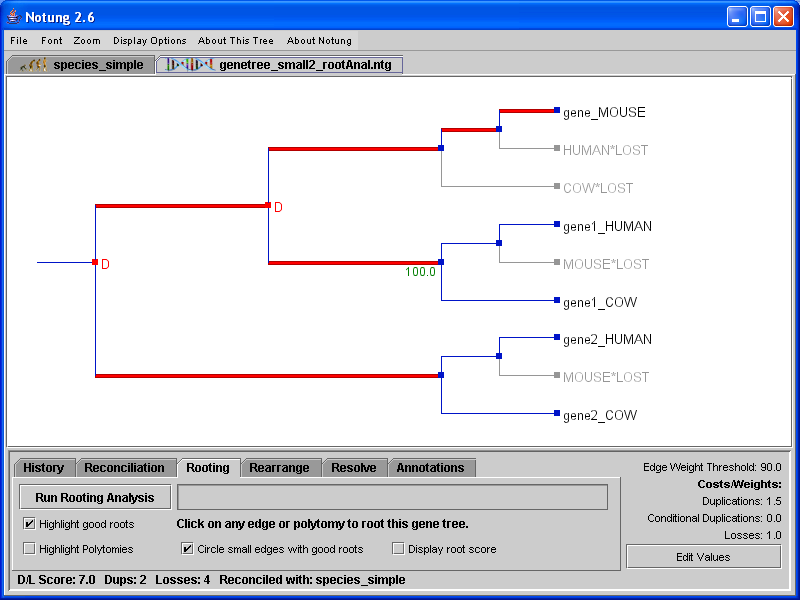
Figure 6.1: (a) The gene tree from Figure 5.1 after completing the Rooting Analysis. (b) The rerooted tree, after the user has clicked on an edge to designate the root.
When the species tree is non-binary, applying Notung’s Rooting analysis to an unrooted, binary gene tree labels the original gene tree with a root score on each edge. This score is a weighted sum of the number of required duplications, conditional duplications, and losses. By default, the cost of conditional duplications is set to zero. Conditional duplications will only influence the root score if this cost is explicitly set to a positive number by the user. For more information on setting parameters, see Chapter 3.5 - Parameter Values.
Rooting analysis when the gene tree is non-binary differs from the binary case in that root scores are assigned to polytomies, as well as edges. Edges and polytomies in the original tree are assigned the D/L Score associated with rooting on that edge or polytomy. If rooting on a polytomy in a non-binary gene tree produces the minimum or near-minimum score, that node will be circled and the vertical edge representing that polytomy will be highlighted in the appropriate color (Figure 6.2).
Click on image to see larger version
Figure 6.2: Rooting analysis for a non-binary gene tree. The optimal root locations are colored in red. If an edge represented by the polytomy can be selected as an optimal root, the polytomy will be circled and colored in red.
To reroot the tree, click on any edge or polytomy in the tree panel. You may root the tree on any edge, not just the highlighted edges. Notung will root the tree on that edge (or polytomy), and recalculate the reconciliation. The D/L Score of the new, rooted tree is displayed in the bottom-left corner of the screen.
Please note that it is not possible to represent an unrooted tree in standard Newick format. Some tree reconstruction programs, therefore, represent an unrooted tree as a rooted tree with a trifurcation (a polytomy with three children) at the root. Notung cannot distinguish between an unrooted, binary gene tree and a rooted gene tree that has a single trifurcation. If such a gene tree is opened and reconciled in Notung, a notification will appear to inform the user that this tree may, in fact, be an unrooted, binary gene tree. Notung will assume that the tree is rooted and non-binary, and will draw the tree and issue diagnostic messages, accordingly. If you consider the tree to be unrooted and binary, you may find this behavior unexpected. If you want Notung to treat the tree as a binary tree, the trifurcation can be removed by rooting the tree in the Rooting panel. The tree can be made binary by manually rooting the tree on any edge; otherwise, the Rooting Analysis may be used to select the edge with the optimal D/L Score.
NOTE: If the tree has not been reconciled before running a Rooting Analysis, Notung will reconcile it automatically. You will be asked to select a species tree for reconciliation (see Chapter 5 - Reconciliation Mode).
To find optimal root edges:
Good roots will be highlighted. If highlighted edges are small, they are circled in the appropriate color to help the user locate them visually. Use the Zoom feature (see Chapter 11.2 - Zoom) to zoom in on these edges.
To show/hide Rooting Analysis results:
In Rooting Mode, the task panel contains several check boxes that allow the user to specify what rooting related information should be displayed.
To reroot the tree:
Weakly-supported edges, as indicated by low edge weights, often imply that the inferred history associated with those edges may not be accurate. Notung can rearrange weakly-supported regions in a gene tree to produce alternate event histories with minimum D/L Score. When these edges or regions are rearranged, the structure of strongly-supported edges or regions stays intact. Any edge that is added as a result of rearrangement will be not be assigned an edge wieght. Since support for edges is determined by edge weight, Notung’s rearrangement function requires that the gene tree include edge weights which assess how well each edge is supported by sequence data. These edge weights can be bootstrap values, probabilities, or branch lengths.
Weak edges are defined as those edges with weights below the Edge Weight Threshold. Selecting the “Highlight weak edges” checkbox in Rearrange mode will highlight all weak edges in yellow, allowing the user to see which edges will be considered for rearrangement (see Figure 7.1). This option is only available in Rearrange mode. The yellow highlighting will disappear when another mode is selected. As a default, the Edge Weight Threshold is 90% of the maximum edge weight. While this is a good starting place for bootstrap values, it may not be appropriate for probabilities or branch lengths. The threshold can be adjusted by the user; see Chapter 3.5 - Parameter Values for information on how to change the Edge Weight Threshold. Notung also considers any edge without an assigned weight to be a weak edge. If Notung’s rearrangement function is applied to a tree with no edge weights, it will consider all edges to be weak, and will find all trees that are optimal when only gene duplication and loss are considered (i.e. those trees with a minimal D/L Score).
Click on image to see larger version[][]
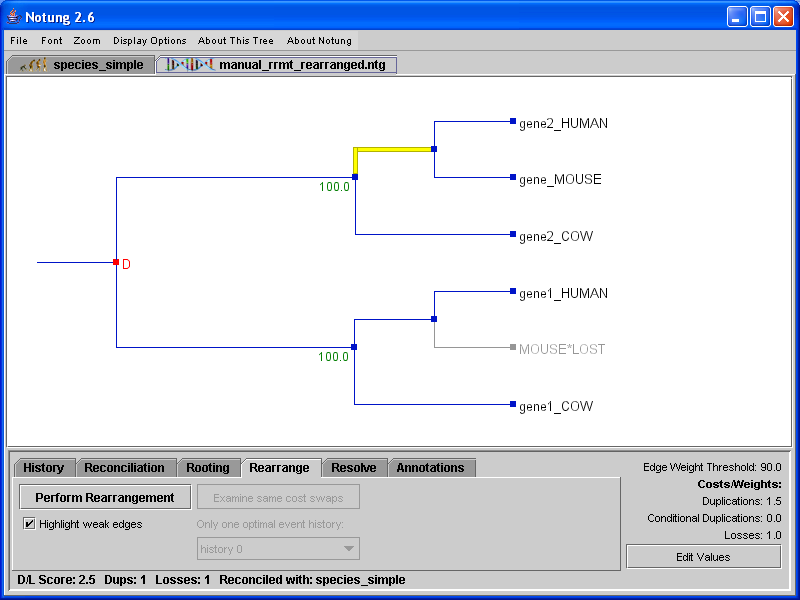
Figure 7.1: (a) The gene tree from Figure 5.1 with weak edges highlighted. (b) After clicking “Perform Rearrangement,” the rearranged tree appears in the tree panel. Weak edges are still highlighted in yellow.
The Rearrangement function can be applied to a non-binary gene tree when the species tree is binary (Figure 7.2). Notung will replace each polytomy with an arbitrary binary resolution, inserting new nodes and edges. These new edges are treated as weak edges. The standard rearrangement algorithm is then applied to the resulting binary tree to determine the rearrangement that results in a minimal D/L Score. Note that it is immaterial how the polytomies are initially resolved, because subsequent rearrangement will result in a minimum cost tree. Rearrangement cannot be performed when the species tree is non-binary.
When rearranging a gene tree, there may be more than one tree that (1) agrees with the original tree at strongly supported edges and (2) has minimal D/L Score. If there are many such trees, considering all of them may be a daunting task. Notung addresses this issue by partitioning the set of all optimal trees into subsets in such a way that any tree in a given subset can be generated from any other tree in the subset by a series of node interchanges.
All trees in any given subset are instances of the same event history. An event history describes a series of events (duplications and losses) and the location in the species tree where they occurred. “A duplication in the common tetrapod ancestor, a loss in the fish lineage and three duplications in mouse” is an example of an event history. To see that more than one tree can have the same event history, note that “three duplications in mouse” corresponds to the subtree ((g1_mouse, g2_mouse), (g3_mouse, g4_mouse)), as well as the subtree ((((g1_mouse), g2_mouse), g3_mouse), g4_mouse).
If multiple minimum cost trees are found, Notung presents one tree from each subset (i.e. one representative of each event history) to the user and provides a point and click interface that allows the user to inspect any other tree in that subset. Initially, Notung arbitrarily selects one event history to present in the tree panel. The other optimal histories may be viewed using the drop-down menu labeled “Select an optimal event history,” which gives a list of up to 50 optimal event histories. The user can perform Same Cost Swaps on a tree to explore the space of all optimal trees corresponding to the current event history. Same Cost Swaps are node interchanges that result in another tree with an optimal D/L score. Clicking the “Examine same-cost swaps” button will highlight all swappable nodes, nodes that can be manually swapped without changing the D/L Score.
If there are more than 50 optimal event histories, they can be generated using the Command Line Interface (see Chapter 12 - Command Line Options and Batch Processing). Note that both the drop down menu and command line options give distinct optimal event histories, but do not generate all optimal gene tree rearrangements. It is only possible to view all trees by performing same cost swaps using the point and click interface in the GUI.
For further details on Notung’s rearrangement algorithm see:
D. Durand, B. V. Halldorsson, B. Vernot. A Hybrid Micro-Macroevolutionary Approach to Gene Tree Reconstruction. Journal of Computational Biology, 13(2): 320-335, 2006.
Click on image to see larger version[][]

Figure 7.2: (a) When rearranging the non-binary gene tree, weak edges are highlighted in yellow. These edges, as well as the polytomies, highlighted in cyan, will be rearranged to produce the binary tree with the minimal D/L Score. (b) After the tree is rearranged, weak edges are highlighted in yellow. Notice that new edges have no edge weight and are considered weak.
To rearrange the gene tree:
A minimum cost rearrangement tree will appear in the tree panel as shown in Figure 7.1(a). Note that weak edges, highlighted in yellow, will not have edge weights. Some or all of these are edges that do not correspond to any bipartition (split) represented in the original tree. The appropriate weights for these edges are not known.
NOTE: If asked to rearrange a tree that has not been reconciled, Notung will reconcile it automatically. In this case, the user is asked to select a species tree for reconciliation.
To highlight all weak edges (default: OFF):
All weak edges in the tree will be highlighted in yellow.
To view alternate optimal event histories:
If more than one optimal event history exists for a rearranged tree, the drop down menu “Select an optimal event history’’ will be enabled.
The tree panel will now show a new tree corresponding to the selected alternate history.
If there is only one optimal history or the tree has not yet been rearranged, the drop down menu will be grayed out.
Click on image to see larger version
Figure 7.3: Swappable nodes are marked with the enlarged square. The selected node, shown in blue, can be swapped with the node highlighted in orange.
Click on image to see larger version
Figure 7.4: Clicking on first the blue node and then on the orange node in Figure 7.3 results in the alternate optimal tree shown here.
To swap individual nodes:
NOTE: If there are no swappable nodes in the tree or if the tree has not yet been rearranged, this button will be grayed out.Swappable nodes are marked with an enlarged blue and cyan square. As you pass the mouse over a swappable node it will be highlighted with a blue triangle. Other nodes that can be interchanged with it with are temporarily highlighted with a light orange triangle, as shown in Figure 7.3. If you have zoomed in, some swappable nodes may be outside the boundaries of the tree panel. Swappable nodes that are not currently visible are indicated by arrows in the tree panel, pointing in the direction of those nodes. These can be seen by scrolling in the direction of the arrow.
NOTE: When a user selects a different alternate event history from the “Select an optimal event history” list, Notung rebuilds the tree from data saved at the time of rearrangement. Any manual swaps made to a previously viewed event history will be lost. Therefore, if you wish to save information after a manual swap, you must save your tree. See Chapter 3.3 - Opening and Saving Trees for more information.
Resolve mode is only applicable to non-binary gene trees. Its function is to resolve polytomies in a non-binary gene tree by comparing it with a binary species tree, resulting in one or more binary tree(s) with minimal D/L Score.
Specifically, the Resolve function removes all polytomies in the original gene tree, and uses an algorithm similar to the rearrangement algorithm to replace them with new edges such that: 1) the new tree is binary, and 2) the new tree has optimal D/L Score. Note that each edge in the original non-binary gene tree still exists in the resulting binary gene tree.
There may be more than one binary tree that agrees with the input tree at all edges except polytomies and has minimal D/L Score. In this case, the user can investigate these optimal alternate hypotheses using a point and click interface as in Rearrange mode. See Section 7.1 - Alternate Optimal Hypotheses for a more detailed explanation of alternate hypotheses.
Selecting the “Highlight Polytomies” checkbox will highlight in cyan all vertical edges representing polytomies in the tree, allowing the user to see which nodes will be resolved. After running the resolve algorithm, the “Highlight New Edges” checkbox will be selected, and will highlight in cyan all those edges in the gene tree that were previously represented by the polytomy (see Figure 8.1). This option is only available in Resolve mode.
Click on image to see larger version[][]

Figure 8.1: (a) Polytomies in the gene tree can be highlighted in cyan while in the Resolve task mode. (b) After the polytomies are resolved, edges that were not present in the original tree are highlighted in cyan.
To resolve the gene tree:
A minimum cost binary resolution of all polytomies in the tree will appear in the tree panel. Note that the new edges will not have edge weights.
If the gene tree is binary, the “Resolve Polytomies” button will be grayed out.
NOTE: If asked to resolve a tree that has not been reconciled, Notung will first invoke the reconciliation algorithm. In this case, the user is asked to select a species tree for reconciliation.
To highlight all polytomies (default: OFF):
All vertical edges representing polytomies in the tree will be highlighted.
To highlight all new edges (default: ON, after resolving):
All edges that were represented by the polytomies in the original tree will be highlighted.
To view alternate optimal event histories:
The tree panel will now show a new tree corresponding to the selected alternate history.
If there is only one optimal history or if the polytomies have not been resolved, the drop down menu will be grayed out.
To swap individual nodes:
NOTE: If there are no swappable nodes in the tree or if the polytomies have not been resolved, this button will be grayed out.Swappable nodes are marked with an enlarged blue and cyan square. As you pass the mouse over a swappable node, other nodes that can be interchanged with it with are temporarily highlighted with a light orange triangle. Swappable nodes that are not currently visible in the tree panel (for instance, if you have zoomed in), are indicated by arrows in the tree panel pointing in the direction of those nodes.
The node you selected is highlighted with a blue triangle. Nodes with which it can be swapped are now highlighted with pink triangles.
NOTE: When a different alternate event history is selected in the “Select an optimal event history” list, Notung rebuilds the tree from data saved at the time of resolution. Any manual swaps made to a previously viewed event history will be lost. Therefore, if you wish to save information after a manual swap, you must save your tree. See Chapter 3.3 - Opening and Saving Trees for more information.
The state of a gene tree changes each time a Notung operation, such as rooting, rearrangement, reconciliation, or resolution, is performed on the tree. Notung maintains a history of state changes for each gene tree. This history can be accessed via the History panel, allowing the user to return to and operate on a previous state, or visually compare the state before and after a task is performed.
Notung lists the states in the history panel by task name (see Figure 9.1). The first entry in the list is always Start, which is the state of the tree when loaded; others entries may include Changed Parameter Values, Reconciled, Rooting Analysis, Rooting on X, Notung Rearrange, Notung Resolve Polytomies, Select Alternate Optimal History, and Swapped Y and Z, where X is an edge and Y and Z are swapped nodes. The list proceeds from top to bottom in the order tasks were performed, and includes the D/L Score for each state.
NOTE: Previous states in the History panel are not saved in a file. When the gene tree file is closed, the history associated with the current tree is lost. To save trees associated with intermediate states, select the state and click “File → Save As.”
NOTE: Parameter values are saved with each state in the history. For each state in the history, the parameters will correspond to those values used at the time the operation was performed. Any subsequent changes to parameter values will not be applied retroactively.
To view previous states of the gene tree:
Click on image to see larger version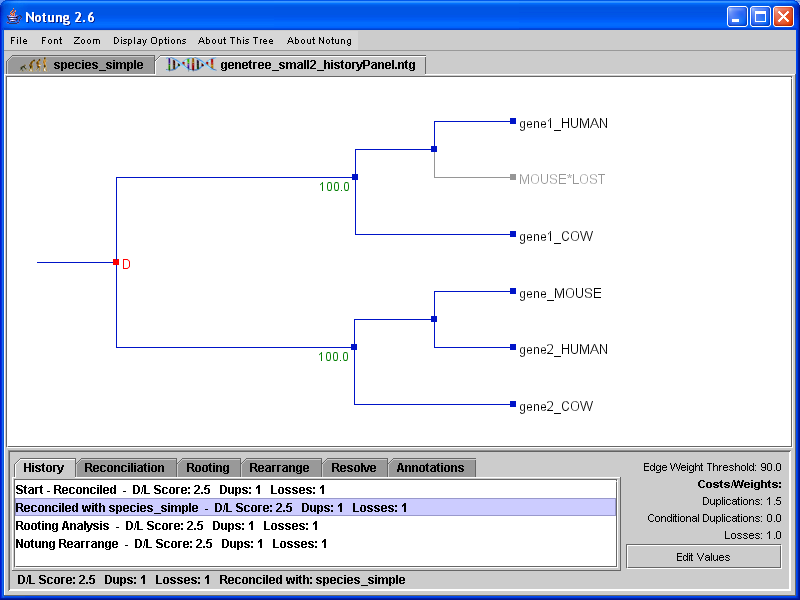
Figure 9.1: The history of a gene tree that had been reconciled, rooted, and rearranged. Currently, the state of the tree after reconciliation and prior to rooting is selected and displayed in the panel.
Notung can annotate the leaf nodes of both gene and species trees with colors specified by the user. For example, the annotation function can be used to color all nodes associated with a particular taxonomic group (e.g., plants) or a particular subfamily (e.g., HSP70). This can help visually differentiate gene clusters in a large and complex tree, or highlight related nodes that are distantly located in a tree.
The “New” button in the Annotations task panel opens the annotations dialog window (see Figure 10.1), where the user can set the annotation parameters. Each annotation consists of a title used to identify it, a color, and a specification of the nodes that are included in the annotation. The title of an annotation is simply an alphanumeric string used to distinguish it. You may use any string of characters as long as it is unique. The set of nodes associated with a given annotation can be specified in two ways, by pattern matching or by selecting them manually. In the first case, the user provides one or more alphanumeric strings, which are compared with all leaf node names. Leaf nodes that contain one or more of the specified strings as a substring are added to the annotation. Alternatively, nodes can be manually added to the annotation by clicking on them.
Click on image to see larger version[][]
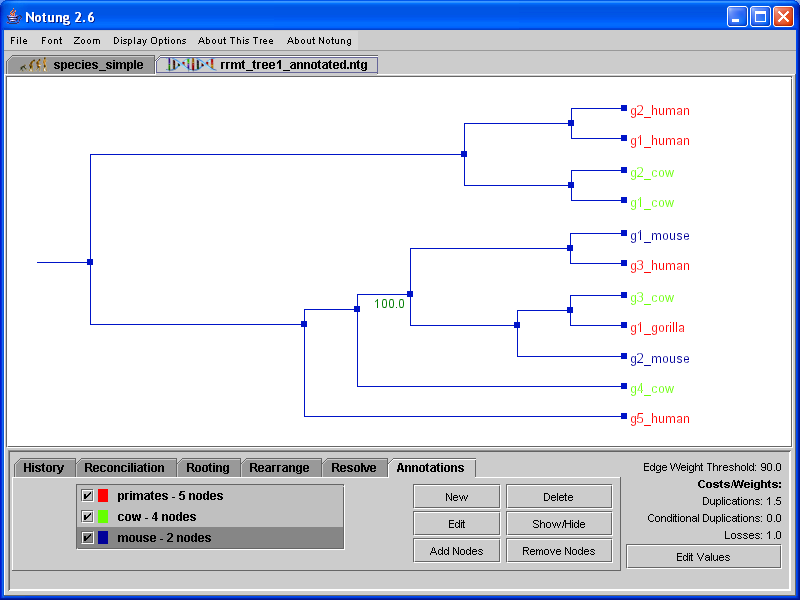
Figure 10.1: (a) The annotations dialog box. This figure shows the creation of an annotation, with the title “primates”, associated with the color red, and the pattern matching terms “hu”. (b) In the Annotations task panel, the list box shows the annotations associated with the currently selected tree. A check box indicates whether the annotation is hidden (unchecked) or showing (checked). The number next to each annotation refers to the number of leaf nodes currently colored by that annotation. If the annotation is hidden, this number is zero.
All annotations for the currently selected tree are shown in the list box in the Annotations task panel (see Figure 10.1b). After an annotation is created, individual nodes can be added to it or removed from it, manually. Annotations can be edited to modify the list of pattern matching terms or to change the color associated with the annotation. Annotations can be shown or hidden at any time.
NOTE: A single node can match more than one annotation, but will only be colored by the most recently created annotation.
NOTE: Annotations only apply to the tree that is currently selected, but can be exported and then imported into another tree. See subsections on importing and exporting annotations at the end of this section.
To create an annotation using pattern matching (recommended):
For example, if you want to annotate all the node labels containing HU, enter “HU” in the text field. Notung will annotate any node with a label that contains “HU” as a substring, such as g1_human and g2_human. If you want to annotate all node labels containing “HU” and “GO,” enter “Hu, Go” in the text field. This will also annotate the node g1_gorilla, as seen in Figure 10.2.
NOTE: This process is not case sensitive.
Nodes with names that match a string in the comma-delimited list will change color (e.g., Figure 10.2).
If a single node corresponds to more than one annotation, the node will be in the color dictated by the most recently added annotation. The newer annotation will continue to take precedence until the shared node is manually removed from that annotation, the annotation is hidden, or a new, conflicting annotation is added. For example, adding an annotation in yellow for “g1” would change the color of g1_human, g1_cow, g1_mouse, and g1_gorilla to yellow.
Click on image to see larger version
Figure 10.2: A fully annotated gene tree.
To create an annotation with manually added nodes:
To add nodes to an annotation manually:
NOTE: This operation can only be performed if an annotation has already been created.
If a selected node is a leaf node, it will be highlighted with the color of the annotation. If it is an internal node, all of leaf nodes below it will be highlighted with the color of the annotation.
To remove nodes from an annotation manually:
NOTE: This operation can only be performed if an annotation has already been created and nodes have been assigned to it.
If a selected node is a leaf node, it will be removed (i.e., disassociated) from the annotation and the color of its label will revert to black (unless an earlier annotation also colors that node). Clicking on an internal node removes all of the leaf nodes in the subtree rooted at that node.
To edit an annotation:
To hide/view an annotation:
If the annotation was displayed prior to clicking the “Show/Hide” button, the nodes associated with the annotation will revert to black (unless an earlier annotation also colors that node). If the annotation was hidden, the associated nodes will appear in color. A check mark next to the annotation’s name denotes that it is visible (i.e., the current state is “Show”). This is the default status.
To delete an annotation:
This function will remove an annotation from the list of annotations. All nodes associated with it will revert to black. Warning: this operation is not reversible.
To export an annotation:
Annotations can be exported to a separate file for import into another tree.
To import an annotation:
Annotations can be imported from any file that contains an annotation, including a Notung format tree or an exported annotation file.
NOTE: Imported annotations are added to the existing list of annotations. If an imported annotation and a previously existing annotation correspond to the same node, the imported annotation will take precedence.
Notung offers the user a broad range of options for controlling the appearance of the tree panel and the types of information that can be displayed. Visual presentation of trees in Notung can be changed in two ways. One set of options is found in the Display Options, Zoom, and Font menus in the upper left hand corner of the Notung window. These options, which are described in this section, are relevant in all task modes.
In addition, certain visual features can be controlled from individual Task Panels. These features are typically specific to that task and, in most cases, are only visible when the relevant Task Panel is selected. These options, such as highlighting swappable nodes or weak edges and displaying root scores, are described in the relevant mode sections.
Notung allows users to show or hide node and edge labels using the Display Options menu. Checkboxes next to each item in the menu show which display options are turned on. These options are tree specific - changing them in the currently selected tree will not change them in other open trees. To turn on/off a display option:
NOTE: If internal nodes are not labeled in the input gene tree file, Notung assigns internal node names using a counting system. Node names derived this way begin with n or r, followed by number signifying the order in which the node was counted (e.g., n136 or r122). Node names beginning with an r indicate that the node did not exist in the original tree; rather, they were added during rearrangement of weak edges or resolution of a polytomy. These node names may change if other tasks are performed after the rearrangement or resolve task that produced the new internal node(s). Counting may be different for each session, so node names may vary from session to session, depending on how many trees have been opened in Notung.
When reconciling a gene tree with a non-binary species tree, if orthologous genes are absent in two or more children of a polytomy in the species tree, a single loss in an ancestral species is inferred (called a polytomy loss). When this option is turned on, polytomy losses are displayed with the names of species in which the gene is absent, as well as name of the polytomy (see Figure 5.2a). When turned off, combined losses are labeled with the number of species in which the gene is absent, and the name and the size of the polytomy (Figure 5.2b.)
Notung allows users to zoom in on the tree using either the Zoom menu or keypad controls. Users can zoom in on the whole tree, maintaining the tree’s aspect ratio, or on the X or Y axis independently, elongating the vertical or horizontal edges, respectively. These changes apply only to the currently selected tree.
To zoom in on the whole tree:
NOTE: Ctrl/Cmd means to use the “Control” key for Windows or Linux, and the “Command” (open apple) key for Mac OS X. For operations which do not involve clicking on the tree, the “Control” key is used for all three platforms.
To zoom out on the whole tree:
To zoom in on the X axis:
To zoom out on the X axis
To zoom in on the Y axis:
To zoom out on the Y axis:
To fit the whole tree in the tree panel:
Users can modify the font size of tree labels using the Fonts menu or keypad controls. Fonts can be set to one of four sizes or changed incrementally.
To set a font size:
To increase font size incrementally:
To decrease font size incrementally:
NOTE: Selecting “Large fonts” does not display the largest possible font; the font can be made even larger by using the “Increase font size” option.
Notung offers a command line interface (CLI) that can perform most operations from the command line without launching the graphical user interface. The CLI allows the use of batch processing to apply Notung to many trees in a large-scale analysis without human intervention. It can also be used to analyze a small number of trees without launching the GUI, for example, by a user executing Notung on a remote computer over the network. The GUI can also be launched from the command line, rather than by clicking on an icon, allowing the user to initiate the GUI with parameter settings other than than the default settings. Finally, when used as an applet, Notung is launched from a web page using CLI syntax.
We follow the following stylistic conventions in this chapter.
-g <genetree>, indicates that Notung expects a file name after
-g.mygenetreefile will produce the file
mygenetreefile.reconciled. In this chapter, we use
<function> to describe such file names, e.g.,
<genetree>.<function>.mygenetreefile.reconciled.0,
mygenetreefile.reconciled.1, etc. We use a ’#’ sign to
represent the number in such file names; e.g.,
<genetree>.<function>.#.Prior to running Notung’s command line interface, you will need to open a command or “terminal” window.
Opening a command window
Click on the Start button, and select the “Run...” item. A dialog box will pop up. Enter “cmd.exe” into the box, and click “OK.”
Navigating to the Notung directory
In the command window, type the followingcd <pathname>where<pathname>is the path of the Notung directory. If the folder location has any spaces in it, it must be enclosed in quotes. For example, if the following is the location of the Notung folder:C:\Documents and Settings\User\Desktop\Notung-2.6Then you should use quotes so that it looks like this in the command window:cd "C:\Documents and Settings\User\Desktop\Notung-2.6"Hit Enter, and you will now be in the Notung Folder.NOTE: To find the path of the Notung directory, select the Notung folder in Explorer, and right click on it. This will pop up a menu - select the Properties item. This will pop up a dialog listing the properties of the Notung folder, including its location.
Opening a command window in the Notung directory
Select the Notung folder in Explorer, and right click on it. This will pop up a menu - select “Start command window here.”
Opening a terminal
The Terminal application is located in the Applications folder in the Utilities subfolder.
Navigating to the Notung directory
In the terminal window, type the followingcd <pathname>where<pathname>is the path of the Notung directory. If the folder location has any spaces in it, it must be enclosed in quotes. For example, if the following is the location of the Notung folder/Users/user/Desktop/New Folder/Notung-2.6Then it should look like this in the terminal windowcd "/Users/user/Desktop/New Folder/Notung-2.6"Hit Enter, and you will now be in the Notung Folder.NOTE: To find the path of the Notung directory, select the Notung folder in the Finder, and select “Get Info” from the File menu. This will pop up a dialog listing the properties of the Notung folder, including its location. You could also drag and drop the Notung folder into the Terminal window to paste the folder’s path into the window.
Navigating to the Notung directory
In the terminal window, type the followingcd <pathname>where<pathname>is the path of the Notung directory. If the folder location has any spaces in it, it must be enclosed in quotes. For example, if the following is the location of the Notung folder/Users/user/Desktop/New Folder/Notung-2.6Then it should look like this in the terminal windowcd "/Users/user/Desktop/New Folder/Notung-2.6"Hit Enter, and you will now be in the Notung Folder.
Notung can carry out its four main tasks, reconcile, rearrange, rooting and resolve, from the command line. In each case, Notung reads in gene and species trees (the input trees) and executes the specified task, resulting in one or more modified trees (the output tree(s)). This modified tree is written to a file. Notung can also generate images in PNG format from the command line. This function can be carried out in conjunction with any of the four main tasks, or independently to generate an image of an existing tree without performing any analysis. The I/O requirements differ somewhat in the latter case; only one tree is required as input and an image rather than a tree file is generated as output. In this section, we discuss executing the four main tasks from the command line, postponing image generation to a later section. In Section 12.3 (Running Notung from a Batch File), automated execution of Notung is described. Commands and options specific to image generation are described in Section 12.4 (Saving PNG Images of Trees). Commands and options specific to reconciliation with non-binary species trees are described in Section 12.5 (Options for Reconciling with Non-Binary Trees).
For the four major tasks, Notung is executed from the command line using the following format:
java -jar Notung-2.6.jar [input tree(s)] [task] [options]
The four main tasks require both a gene tree and a species tree. These are usually supplied as two separate input files. A single file containing a previously reconciled tree in Notung format is also acceptable, since such files contain both a gene tree and species tree. If a gene tree file containing a reconciled tree in Notung format and a species tree in a separate file are both given, the latter is used; the species tree in the gene tree file is ignored. The task parameter must be one of --reconcile, --rearrange, --root, and --resolve (the fifth task, --savepng, is discussed in Section 12.4.) Options are described below.
NOTE:
- The input trees, tasks, and options may be given in any order.
- To launch the graphical interface from the command line, run Notung with no task option.
- Running Notung with the --help option causes it to print information regarding input, output, and other options.
- The commands given in this chapter will only work if you are currently in the same directory as the Notung jar file. In order to run Notung from any directory, add the Notung directory to your CLASSPATH. For example, if you run bash In Linux, you can do this by adding the following command to your .bashrc file:
setenv CLASSPATH $CLASSPATH:<pathname>See a java manual for more information about CLASSPATH settings.
The following list describes Notung’s command line options. For more details on tree formats, including information on edge weights, species tags and output files, see Appendix A - File Formats.
If one of the four main functions is given, the output gene tree
will be saved to a file called <genetree>.<function> (where
<function> is one of the four major tasks, reconcile,
rearrange, resolve, or rooting.) If the analysis results in more
than one optimal history,
then the output files are
numbered, (e.g. <genetree.rearrange.0,
<genetree.rearrange.1, etc.).
By default only one tree is saved. To
save more than one tree, use --maxtrees.
If the --savepng option is given, an image of the tree is saved in PNG format. For more information on saving PNG images with --savepng, see Section 12.4 - Saving PNG Images of Trees.
If the species tree contains species that do not appear in the gene
tree, during reconciliation Notung constructs a pruned species tree
that only contains those species required to reconcile the gene
tree. If the --stpruned option is given, this pruned
species tree is saved in the file
<genetree>.<function>.species.
When run on the command line, Notung outputs status information to
the terminal window. This information can be saved in the log file
<genetree>.<function>.ntglog by using the --log
option. For a batch run, a log file is not saved for each tree;
rather, a single log file for the entire batch run is saved to the
file <batchfile>.<function>.ntglog.
General tree statistics can be saved in the file
<genetree>.<function>.stats by giving the option
--treestats. This file includes information on both the gene
tree and the pruned species tree. For more information on tree
statistics, see Section 3.4 - General Tree Statistics.
Information on the timing of
each duplication and loss is saved in the file
<genetree>.<function>.info when the --info option
is used.
For each duplication, an upper
and lower bound (represented as nodes from the species tree) are
given. For losses, each node in the species tree is listed with the
number of losses associated with that taxon. For more information
on duplications and losses, see Chapter 5 - Reconciliation Mode.
Notung can output tables of orthologs and paralogs for all pairs of leaf nodes in the reconciled tree. This table can be generated in several formats: comma-separated values (CSV), tab-delimited values, or an html-formatted table. Use options --homologtablecsv, --homologtabletabs or --homologtablehtml, respectively. For more information on orthologs and paralogs, see Section 5.3 - Inferring Orthologs and Paralogs.
Load the file <genetree> as a gene tree.
NOTE: The -g is optional.
Load the file <speciestree> as a species tree.
The -s is required.
Load the trees listed in <batchfile>. Requires that the
--speciestag option be set. If rearranging, requires the
--edgeweights and --threshold options. With
this option, -g <genetree> and -s <speciestree>
should not be specified. See Section ?? - Running Notung from a Batch
File for more information.
Files listed in <batchfile> use absolute paths.
See Chapter 12.3 - Running Notung from a Batch File for more information.
Load gene tree from a URL. This option is only used when running Notung as an applet.
Load species tree from a URL. This option is only used when running Notung as an applet.
Reconcile a gene tree with a species tree. In batch mode, --speciestag is required. For more information on reconciliation, see Chapter 5 - Reconciliation Mode.
Rearrange the gene tree. The option --threshold must be set. In batch mode, --speciestag and --edgeweights are also required. For more information on rearranging gene trees, see Chapter 7 - Rearrange Mode.
This task, which removes polytomies from a non-binary tree, can only be carried out if the gene tree is non-binary. In batch mode, --speciestag is required. For more information on resolving non-binary nodes in a gene tree, see Chapter 8 - Resolve Mode.
Root the gene tree. The top <maxtrees> best scoring
rooted trees are saved in files named
<genetree>.rooting.#. By default, <maxtrees> is
set to 1. In batch mode, --speciestag is required.
For more information on rooting gene trees, see Chapter 6 - Rooting Mode.
Sets the cost of gene duplications. If not set, the cost is set to 1.5, by default.
Sets the cost of conditional gene duplications. These only occur when reconciling a binary gene tree with a non-binary species tree. If not set, the cost is set to zero, by default. See Chapter 5 - Reconciliation Mode for more information.
Sets the cost of gene losses. If not set, the default cost of 1.0 is used.
Indicates the format of species tags in the gene tree. If not set, Notung tries to guess the correct format. See Appendix A.4 - Specifying the Species Associated with Each Gene.
Edges with weight higher than <threshold> are preserved
during rearrangement. This can be given as an absolute value or
or as a percentage of the maximum value, using <percentage>%;
e.g. “--threshold 90%” sets the threshold at
90 percent of the highest edge weight in the tree.
See Section 3.5 - Parameter Values for more information.
Indicates where in the tree file the edge weights, if any, are specified. If this option is not set, and the gene tree has values in more than one location, Notung will guess the location of edge weights when using --rearrange. See Appendix A.6 - Location of Edge Weight Values for more information.
Same setting as --edgeweights. Kept for backwards compatibility.
Attach the given annotation file to each input tree.
Used with --savepng. Notung uses the contents of
<filename> to create an image map file, which is saved in
<outputtreename>.png.html. For more information,
see Section 12.4 - Saving PNG Images of Trees.
Specify output tree file format. See Appendix A - File Formats for more information.
Remove loss nodes from gene trees before they are saved. Useful when outputting tree in Newick or NHX formats, which do not recognize loss nodes, or with --savepng to output a tree image without loss nodes.
Maximum number of optimal trees to output during reconciliation, rearrangement, rooting, and resolving. Default is one.
Save output files in the
directory, <outputDir>. Default is the current
working directory.
Save output trees in the directory in which
<genetree> is located.
Writes diagnostic output to the file
<genetree>.<function>.ntglog, where <function> is
one of the four modes. For batch runs, the log file is saved in
<batchfile>.<function>.ntglog.
Save information on duplications and losses in the
file <genetree>.<function>.info.
Save general statistics for a tree. Saved in
<genetree>.<function>.stats. Statistics on the
pruned species tree will be included in this file.
See Section 3.4 - General Tree Statistics for more information.
Save a version of the species tree that
contains only the species found in the gene tree. Saved in the
file <genetree>.<function>.species.
Report a list of ordered root scores to standard output (only used with --root). This option is useful for statistical examination of root scores for the gene tree. These scores can be saved in a file with the --log option.
Suppresses reporting of diagnostic information to the terminal.
In batch mode, print a simple progress bar to stderr for each tree analyzed. Useful with –silent.
Save the tree as a PNG image. Unlike Notung’s other main functions, this function does not require a species tree. For more information about --savepng, see Section 12.4 - Saving PNG Images of Trees.
For more information on orthologs and paralogs, see Section 5.3 - Inferring Orthologs and Paralogs.
Save a comma separated table of orthologs and paralogs to the file
<genetreename>.<function>.homologs.csv.
Save a tab-delimited table of orthologs and paralogs to the file
<genetreename>.<function>.homologs.tabs.
Save a table of orthologs and paralogs in html format to the file
<genetreename>.<function>.homologs.html. This format
can be included in a a web page.
GUI only: if an input gene tree is reconciled, open the attached species tree in a separate tab. Useful for displaying Notung format trees in the Notung applet.
GUI only: if an input gene tree is reconciled, start Notung in the Reconciliation tab with the Orthologs/Paralogs button selected. Useful for ortholog / paralog analysis in the Notung applet.
Print information about these options.
Batch processing allows the user to apply Notung to many trees in a large-scale, automated analysis. The input trees are given in a batch file, which consists of a list of tree file names, one per line. Blank lines and lines which start with # are ignored.
NOTE: By default, Notung expects the tree file locations to be given relative to the location of the batch file. For example, if the batch file is in /username/batchRun, Notung expects the gene trees to be in the batchRun folder or in some subfolder of batchRun. Use the --absfilenames option to indicate that file names are absolute path names.NOTE: When using the --savepng option without any of the four main functions (--reconcile, --root, --rearrange and --resolve), each tree listed in the batch file is saved as an image. For more information, see Section 12.4 - Saving PNG Images of Trees
A sample batch file is provided with the Notung 2.6 distribution in the sampleTrees/batch directory. This batch file includes all combinations of binary and non-binary gene and species trees. Because not all of Notung’s task modes work for each of these combinations, you will receive one or more warnings and errors when running this batch file. In addition, the batch file lists a gene tree which does not exist, to give an example of the appropriate warning.
Use the -b <batchfile> option.
For example, from the Notung directory, enter the following on the command line:
java -jar Notung-2.6.jar -b sampleTrees/batch/batch.run --reconcile --speciestag prefix
The --reconcile option tells Notung to reconcile all the gene
trees listed in batch.run with the species tree listed in
batch.run. The --speciestag prefix option tells
Notung how species labels are specified in the gene tree files, and is
required in batch mode. See Appendix A.4 - Specifying the Species Associated with Each Gene for more information on species labels.
NOTE: All gene trees in the same batch file must use the same species tag format, which is specified using the --speciestag option.
In batch mode, the --speciestag option is always required. In addition, when using --rearrange, --edgeweights and --threshold must be used to set the edge weight locations and threshold, respectively.
As Notung reads and processes each gene tree in the batch file, it prints diagnostic information to the terminal. Notung will also print this information to a log file when the --log option is given. Any errors that occur in the processing of a batch file are reported to the terminal as they occur. The total number of errors is reported at the end of the batch run.
To print status information to a file:
Use the --log option from the command line. The information
will then be written to the file
<batch_file_name>.ntglog.
To save trees to a different directory:
By default, Notung saves each reconciled tree to the directory from which the program was run.
--outputdir <outputDir> option from the
command line. The information will then be written to the
directory <outputDir>.
Progress Bar
For long runs, it may be convenient to use the options --silent and --progressbar together. This will suppress all output to the terminal with the exception of a simple progress bar to stderr. The option --log can still be used to save the (now suppressed) output to a file.
The option --savepng saves a simple image representation of a tree in PNG format. The option --savepng can be used with one of the four main tasks (--reconcile, --root, --rearrange and --resolve), in which case an image of the final output tree is saved, in addition to the output tree file. This behavior is similar to other output options such as --treestats and --homologtablecsv. Alternatively, --savepng can be used alone to save an image of a tree without performing any other tasks.
When --savepng is used without one of the main four tasks, Notung reads in a tree and generates and saves an image of that tree in PNG format. Unless a batch file is used, only a single tree can be processed at a time (i.e., a gene tree and a species tree cannot both be given). If the input tree is a previously reconciled tree in Notung format, the image will show the appropriate duplications and losses (to save an image without losses, use --nolosses). If the tree has not been reconciled, the tree image will show only the structure of the tree and the names of the leaves of the tree.
When using a batch file, each tree specified in the file is saved as an image. When generating images without performing a major task, the batch file format format differs slightly: Species trees and gene trees can be listed in any order.
When --savepng is used alone, an image of the input tree is saved in the file
<treename>.png. When used with --reconcile,
--root, --rearrange or --resolve, an image
of the output tree is saved in the file
<genetreename>.<function>.png. For analyses with more than one
optimal history, an image file is saved for each history. The
number of files is limted by the parameter --maxtrees.
If a tree in Notung format contains color annotations, the leaves in images of that tree will be colored as specified by those annotations. Additionally, an annotation file can be specified with the option --annotationfile. For more information on color annotations, see Chapter 10 - Annotations.
Notung provides the option to produce an html imagemap for a tree
image. If an imagemap and image file are both included in a web page,
each gene in the image will provide a link to a specified web page.
The format of these links is determined by the imagemap specification
file given with --imagemapfile <imagemapfilename>, described below.
The resulting imagemap is saved in the file
<outputtreename>.png.html, where <outputtreename> is
either <genetree>.<function> or <treename>.
To include the image and imagemap in a web page, insert the entire
contents of the saved imagemap file into the html of the web page.
The saved image must be in the same directory as the web page,
unless you specify a different location for the image by changing
<imagefile> in the line:
<img border=0 src='<imagefile>' ...
The specification file given by --imagemapfile <imagemapfilename>
consists of a list of gene/link pairs. Blank lines and lines that
start with # are ignored. An example specification file:
# Danio rerio links: gene: Danio_rerio|(id) link: http://zfin.org/cgi-bin/ZFIN_jump?record=(id) # generic imagemap - everything else links to google gene: (id) link: http://www.google.com/search?q=(id)
Lines starting with ‘gene:’ match genes in the gene tree; lines starting with ‘link:’ specify the format of links for those genes. For each gene in the gene tree, the first gene/link pair that matches will be used. If a gene does not match any of the ‘gene:’ lines, a warning will be printed.
The identifier ‘(id)’ will match any text string, and that
text string is used in the link. Any other text present in the
‘gene:’ line must match gene names exactly. In the example
above, the gene Danio_rerio|ZDB-GENE-031007-1 would match
the first ‘gene:’ line. The identifier (id) would
be ZDB-GENE-031007-1, and the link would be
http://zfin.org/cgi-bin/ZFIN_jump?record=ZDB-GENE-031007-1.
The gene Homo_sapiens|gene1 would match the second pair,
because ‘(id)’ will match any text string. The resulting
link would be
http://www.google.com/search?q=Homo_sapiens|gene1.
An example gene tree and imagemap specification from the Princeton Protein Orthology Database (http://ortholog.princeton.edu/) are included in the Notung distribution.
When inferring losses during reconciliation with a non-binary species tree, it is not possible to determine unambiguously the edge in the the gene tree to which a loss should be assigned. Notung uses two different methods to deal with this problem. An exact algorithm finds all possible assignments that minimize the total number of losses but has exponential time complexity. A heuristic, which runs in polynomial time, is not guaranteed to find the optimal assignment, but usually does in practice. These issues and algorithms are discussed in detail in Section 4 (Non-Binary Trees).
Only the heuristic is implemented in the GUI. Either method may be used when executing Notung from the command line. The CLI runs the heuristic by default. To use the exact algorithm, include the --exact-losses option when running Notung from the command line with the --reconcile or --root tasks.
The running time of the exact algorithm is exponential in the size of
the largest polytomy. Even when --exact-losses is used,
Notung does not apply the exact algorithm to polytomies with more than
12 children. Instead, the heuristic is applied to these polytomies.
To change the maximum polytomy size for which Notung uses the
exact algorithm, use the --polytomy-cutoff <maxPolytomySize>
option when including the --exact-losses option in the
command line.
NOTE: Changing the polytomy cut-off to a larger value and using the exact algorithm on a species tree with a polytomy with more than 12 children may greatly increase running time.
Computes the minimum number of losses when reconciling a binary gene tree with a non-binary species tree. If this option is not included on the command line, the heuristic used. NOTE: In Notung 2.5, this option was named --combine-losses.
Using this option with --exact-losses will change the
default value for polytomy cut-off. Only for losses associated
with polytomies less than or equal to <maxPolytomySize>
will the exact algorithm be used. The default value is 12. If
a polytomy greater than <maxPolytomySize> is encountered,
a warning will be printed to the terminal window and/or log
file.
When run with --exact-losses, this option will report both the number of losses obtained with the heuristic and with the exact algorithm. This is useful for determining whether the heuristic is overestimating the number of losses and by how much. NOTE: In Notung 2.5, this option was named --report-explicit-losses.
Notung can save trees in three different file formats: Newick file format, NHX file format, and Notung file format.
Newick file format specifies tree topology and node labels, but cannot be used to save reconciliation information or information about the species tree with which the gene tree was reconciled.
NHX and Notung file formats use the Newick comment field to store additional information not captured in the standard Newick specification. A reconciliation involves a gene tree, a species tree, the mapping from gene tree to species tree, and the inferred duplications and losses. Newick format stores only the gene tree. NHX format can store a gene tree, with additional information to indicate which nodes are duplications. Notung file format can store a gene tree, the species tree with which it was reconciled, and duplication and loss nodes. If you save a reconciled tree in Notung format, it will still be reconciled when you next open it in Notung.
The Notung file format holds more information, but may not be compatible with other software packages that use Newick format. The formal specification of Newick file format allows bracket-delimited comments. Programs that follow the formal specification and ignore information stored in comments will be able to read NHX or Notung format trees. However, not all programs allow comments. If you plan to use a program that does not allow Newick comments to further analyze trees saved by Notung, save your trees in standard Newick format.
Newick is widely used by phylogeny programs. PHYLIP [], PAUP* [], and many other programs will output trees in Newick.
The general Newick syntax looks like this:
treefile → subtree;subtree → descendant_list [internal_node_label] [:branch_length]
descendant_list → (subtree, subtree [, subtree]) | leaf_node_name
where descendant_list is a string that specifies the
organization of the subtree and
internal_node_label is the
label of the root of a subtree. The optional branch_length
field refers to the length of the edge from the root of the subtree to
its parent. The internal_node_label and
branch_length fields are optional. Some programs use these
fields to store other information. For example, Notung allows the
user to use either of these fields to store edge weight values.
Comments in Newick format are enclosed in square brackets and may appear anywhere newlines are permitted. Some programs use the comment field to store additional information that is not included in the Newick specification. By convention, this information is formatted as follows:
[&&ApplicationID:Application_specific_comments]
where ApplicationID indicates a specific program or format.
For more information about Newick file format, go to:
http://evolution.genetics.washington.edu/phylip/newicktree.html.
or
http://geta.life.uiuc.edu/~gary/Newicks\_845\_Tree\_Std.html.
NHX File Format is based on the Newick file format, but embeds additional information about each node in the tree in the comment fields, as follows:
[&&NHX:TagID1=value1:TagID2=value2]
where TagID1 and TagID2 can specify bootstrap values, species labels, or duplication information. This example has two tags, but NHX comments can have one or more tags. Trees saved in NHX file format include information produced by a reconciliation, including duplications and species labels, but do not record any visual annotations made in Notung. Nor do they record the species tree with which the gene tree was reconciled.
NOTE: The NHX format is case-sensitive.
More information about NHX format, including a complete list of tags used in comment fields, can be obtained at:
http://www.genetics.wustl.edu/eddy/forester/NHX.html.
Notung File Format further extends the NHX format. Notung file format can record duplication marks, edge weights, and color annotations. A reconciled gene tree file saved in Notung format will also have a pruned species tree embedded in it. When the reconciled gene tree is reopened in Notung, the pruned species tree can be extracted and used in the same way as any other species tree. A reconciled gene tree saved in Notung file format also stores additional information on parameter values, including edge weight threshold, loss cost, duplication cost, and conditional duplication cost. In addition, a non-binary gene tree reconciled with a binary species tree with more than one optimal history stores information regarding which history was displayed when saved. When the gene tree is reopened in Notung, the tree for that optimal history will be displayed.
To open an embedded species tree in a Notung format gene tree file:
NOTE: None of the three file formats used in Notung embed alternate histories for gene trees discovered through rearrangement. When saving after rearrangement, Notung saves only the history that currently appears in the tree panel. To access the other alternate histories when opening such a file, the tree must be rearranged again in Notung.
In order to perform reconciliation, Notung must determine the species from which each leaf taxon in the gene tree was derived. This is achieved by embedding the species name in the gene leaf label or by using information embedded in the NHX comment field.
Notung offers three different conventions for specifying the gene to species mapping, described below. Notung will attempt to guess the naming convention used; you can also specify this in the reconciliation dialog (see Chapter 5 - Reconciliation Mode).
NOTE: When using this format, no species label should be a prefix of another species label, such as with carp and carpinusBetulus. In this situation, Notung may incorrectly identify the gene carpinusBetulus_gene1 as a carp gene, rather than a hornbeam gene.
NOTE: Postfix mode cannot be used if species names include underscores (_); for example, Carpinus_betulus cannot be used in Postfix mode.
In previous versions of Notung, punctuation (-, /,
_, ., \) in species names was used to indicate
that Notung should look for a shorter species tag in gene names,
rather than looking for the entire species name. For example, given
the species name Hu.Homo_Sapiens, Notung would look for the
species label “Hu” in gene names.
Because many users found this confusing, this functionality has been
removed in Notung 2.6. Notung now looks for entire species names
during reconciliation, which also allows users to use species names
like Pan_troglodytes and Pan_paniscus in the same tree
without creating a conflict. Unfortunately, this means that some
trees that were used in previous versions of Notung will not work in
the current version. This section explains how to change these
trees so that they can be used with Notung 2.6.
Any species tree with punctuation in the species names, where the full species names are not present in either the gene tree names or in NHX style species tags, will need to be converted. If your species names contain punctuation and you used them with older versions of Notung, then your trees probably fit this description. If Notung 2.6 is used to open an older Notung format tree that needs to be converted, a warning dialog will be shown.
There are three ways to convert trees with punctuation in species names. The correct method to use depends on your desired outcome.
Hu-gene01”,
change “Hu.Homo_sapiens” to “Hu” in the species
tree. These shorter species names should now match the species
labels in the gene names.Hu-gene01” to “Hu.Homo_sapiens-gene01” in the
gene tree. This solution will not work in
Postfix mode if your species names contain underscores (_).If the gene tree is already in NHX or Notung format, modify
the NHX comment after each gene name.
To modify an existing NHX comment, find the species tag and replace
the shorter species label with the full species name. For example,
“[&&NHX:S=Hu]” becomes
“[&&NHX:S=Hu_Homo_sapiens]”.
If there are no comments in the file (i.e., the tree is in Newick
format), add the following after each gene name:
“[&&NHX:S=<speciesname>]”, where <speciesname> is
the corresponding full species name from the species tree. For
example, the gene tree:
(gene1_Hu,
(gene2_Hu, gene2_Mu));
would become:
(gene1_Hu[&&NHX:S=Hu_Homo_sapiens],
(gene2_Hu[&&NHX:S=Hu_Homo_sapiens], gene2_Mu[&&NHX:S=Mu_Mus_musculus]));
Notung uses edge weights to determine which edges are weakly supported and may be rearranged. These edge weights may correspond to bootstrap values, probabilities, branch lengths, or any other numerical indication of support.
Edge weight values can be located in one of three places in a tree file, depending on how the file was created. In Newick format, either the branch length field or the internal node name may be used to specify edge weights. Many programs store bootstrap values in the Newick node name field. In an NHX or Notung format file, edge weights can also be specified using the NHX bootstrap tag in the comment field.
The example below shows a tree with a single edge weight in each of the three tree formats:
(cow_gene1, (mouse_gene2, cow_gene2):100)
(cow_gene1, (mouse_gene2, cow_gene2)100)
(cow_gene1, (mouse_gene2, cow_gene2)[&&NHX:B=100])
Confusion can arise if an input tree has edge weights in more than one type of field. This could occur, for example, in a tree that has both branch lengths and bootstrap values. Notung tries to guess the type of edge weight specification in the file, but it is not always possible for Notung to determine this unequivocally. You can specify the location explicitly using command line options (see Chapter 12 - Command Line Options and Batch Processing) or using the “Select Location of Edge Weights” dialog in the Display Options menu (see Figure A.1).
Click on image to see larger version
Figure A.1: The “Select Location of Edge Weights” dialog box.
To set the location of edge weights in Notung:
The gene tree will immediately reflect the change, so you can check the tree panel to verify that the choice you selected gives the desired values.
Most functions in Notung require a species tree. If you are familiar with the species in your data set, you may already have an appropriate species tree. If you do not have one, you can construct one using resources available on the web.
One such resource is the NCBI Taxonomy Browser, available at the NCBI website:
http://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi
The Taxonomy Browser contains a database of all organisms represented in the NCBI sequence database, and can automatically build a species tree using species selected by the user. To create a tree in a format Notung can understand, add the species to be included in the tree, and then use the Taxonomy Browser’s “Save As” option to save the tree as a Phylip tree. The Phylip option causes the tree to be saved in a variant of Newick format. The resulting tree can then be loaded into Notung as a species tree.
NOTE: The Taxonomy Browser does not recognize all common species names. Formal names for species can be found at:
http://www.expasy.org/cgi-bin/speclist
To build a species tree using the NCBI Taxonomy Browser:
Additional resources provide access to existing species trees built by other researchers. TreeBASE (http://www.treebase.org/treebase/search.html) allows users to search for species trees from a large database of published papers. The Angiosperm Phylogeny Website and the Phylomatic Project provide species trees for plant species.
http://www.mobot.org/MOBOT/research/APweb/welcome.html
http://www.phylodiversity.net/phylomatic/phylomatic.html
Other tree-building tools are listed on Felsenstein’s Phylogeny Programs website:
http://evolution.genetics.washington.edu/phylip/software.html.
NOTE:
Appendix E Worked Examples
Key Combination Action Ctrl + O Open a gene tree Ctrl + Shift + O Open a species tree Ctrl + S Save the tree Ctrl + P Print the current view Ctrl + Shift + R Reload tree from file Ctrl + W Close tree Ctrl + = Increase font size (for all labels in the tree) Ctrl + - Decrease font size (for all labels in the tree) Ctrl + click on tree Zoom in on tree Shift + click on tree Zoom out of tree Ctrl + ] Zoom in on tree on the X-axis Ctrl + [ Zoom out of tree on the X-axis Ctrl + Shift + ] Zoom in on tree on the Y-axis Ctrl + Shift + [ Zoom out of tree on the Y-axis Ctrl + T Show whole tree Ctrl + . Go to next tree Ctrl + , Go to previous tree Ctrl + Q Exit (end Notung)
NOTE: Ctrl indicates use of the control key. Ctrl + click on tree means that the user needs to click on the tree while pressing the appropriate key. Mac users may have to use the command, or open apple key to zoom in on the tree (i.e., command + click on tree), but should use the control key for all other operations.
The following exercises will help familiarize you with the basic tasks Notung can perform on a gene tree. The tree files used in these exercises are included in the Notung distribution, in the sampleTrees folder. If the program window becomes too cluttered, you may close trees that are no longer being used by selecting the tree and clicking on “File → Close.”
In this exercise, you will reconcile the gene tree genetree_NOTCH with the species tree speciestree_mega. You will also generate a pruned species tree, and use Notung to determine the upper and lower bounds on the time when a duplication occurred.
Open the tree files
The gene tree is located in the sampleTrees folder, which is included in the downloaded zip file. Once loaded, the gene tree is displayed in the tree panel.
The species tree is located in the sampleTrees folder. Once loaded, the species tree appears in the tree panel. Because it is the most recent tree opened, it is now selected.
Note that the options that Notung offers differ depending on whether a species tree or a gene tree is selected. For example, because speciestree_mega is now selected, the box showing parameter values in the lower right corner has disappeared, and the task panel includes only two task modes, History and Annotation.
Reconcile the gene tree with the species tree
The Reconciliation task panel opens below. From here you can reconcile a gene tree with a species tree, display a pruned species tree, show duplication bounds, and hide duplication marks and loss nodes.
The Reconciliation dialog appears. In this dialog box, Notung asks you to specify which species tree to use for the reconciliation and what naming convention is used in the gene tree to specify the species associated with each gene.
Currently, the only selection available is speciestree_mega. However, if you have more than one species tree open in Notung, you must specify here which species tree to use.
This section in the dialog box asks you to specify the naming convention used in the gene tree to indicate from which species the genes originated. Notung tries to guess the naming convention, but it does not always guess correctly. Notung should have guessed correctly in this case. In general, remember to check the leaf node names in your gene tree during this step to make sure that they agree with the naming convention you choose.
For more details about the species label naming conventions, see Appendix A.4 - Specifying the Species Associated with Each Gene.
Click on image to see larger versionThe reconciled gene tree now appears in the tree panel. The D/L Score of the reconciled tree, displayed in the bottom-left corner of the program window, is 20.5 - five duplications and thirteen losses. Five red D’s in the tree mark the inferred duplications. At the right end of the tree (at the leaves), thirteen loss nodes appear in light gray type.
Figure E.1: The gene tree should now look like this.
Display the pruned species tree
The leaves of speciestree_mega include more species than are relevant to genetree_NOTCH. After reconciliation, you can view the species tree pruned of all species that are not represented by genes in the gene tree.
A dialog box appears asking you to give a title for the pruned species tree. The default title is “Pruned Species Tree.”
The pruned species tree appears in the tree panel. It contains only seven leaf nodes, all of which are species represented in the reconciled gene tree. The pruned species tree has a tab above the tree panel, labeled “Mega_Pruned.” You can now select and use this tree as you would any other species tree.
Click on image to see larger version
Figure E.2: The pruned species tree should look like this.
Check the duplication bounds
The duplication bounds provide information regarding when gene duplications occurred in the course of species evolution.
Node name labels appear in red type next to each internal node. You can now identify each duplication by name. If internal node names are not provided in the gene tree file, Notung will assign the node an alphanumeric name (e.g. n132).
Node name labels appear in red type next to each internal node.
A new window appears. Inferred duplications are listed in the left column, expressed as node names in the gene tree. The lower and upper bounds are listed in the middle and right columns, respectively, and are expressed as internal node names in the species tree. Information on losses is displayed below duplication bounds. The left column lists the species nodes in the species tree. The right column provides the number of losses that occurred in each species.
The node name may vary, depending on how many internal nodes Notung has counted in your current session.
With Mega_Pruned selected, you can see internal nodes representing euteleostomi and coelom. The duplication occurred somewhere on the edge between those nodes.
The gene tree genetree_ANK is unrooted. In this exercise, you will select a root based on duplication loss parsimony.
Open the tree files
The gene tree is located in the sampleTrees folder.
Since this tree is unrooted, it has a trifurcation (a node with 3 children) at the top of the tree, but is otherwise binary.
Run the Rooting Analysis
The Rooting task panel is displayed. Notung is now in Rooting mode.
A diagnostic message appears warning you that this tree contains a trifurcation at its root and may be unrooted. Click “OK.”
You will be asked to reconcile the tree. Select speciestree_mega and “Prefix,” click “Reconcile”. The edge at the top of the tree panel, leading to caeel*unc-44, is colored red. This means it has the minimum root score.
Each edge is labeled with its root score. Notice that the red edge leading to caeel*unc-44 has a root score of 4.0. The next lowest score is 8.5.
Select a root
The tree is now rooted on the edge leading to the caeel*unc-44 gene. The D/L Score of the tree is now 4.0, with two duplications and one loss.
Click on image to see larger version
Figure E.3: The gene tree should now look like this.
In this exercise, you will reconcile the gene tree genetree_SMALL with the species tree speciestree_small and use Notung’s rearrangement tasks to investigate alternate gene trees with minimum D/L Score. Both input trees are located in the sampleTrees folder.
Reconcile the gene tree with the species tree
This is an artificial tree made up for this exercise. The edge weights in this tree represent bootstrap values. Note that two internal edges have a bootstrap value of 100, one has a bootstrap value of 73, and several have not been assigned a weight. (Note that edges adjacent to leaves are usually not assigned bootstrap values since those edges are present in all trees.) Notung sets the default edge weight threshold to 90% of the maximum edge weight in the tree. Since the maximum edge weight in this tree is 100, the edge weight threshold is set to 90.0.
The reconciled tree appears in the tree panel. Note that it has a D/L Score of 10.0, with four duplications and four losses.
Rearrange the reconciled tree
The Rearrange task panel is now displayed.
Several edges in the reconciled tree are highlighted in yellow. These are edges with weights below the Edge Weight Threshold and are considered “weak.” Weak edges may be rearranged to reduce the number of duplications and losses in the tree. Edges with weights above the threshold will not be rearranged.
Note that in addition to the edge with weight 73.0, the internal edges with no edge weight are also highlighted in yellow. Notung assumes that any internal edge that is not explicitly assigned a weight is considered weak.
Click on image to see larger version
Figure E.4: The gene tree with weak edges highlighted.
The rearranged tree appears in the tree panel. It now has a D/L Score of 4.0, with two duplications and only one loss.
Click on image to see larger version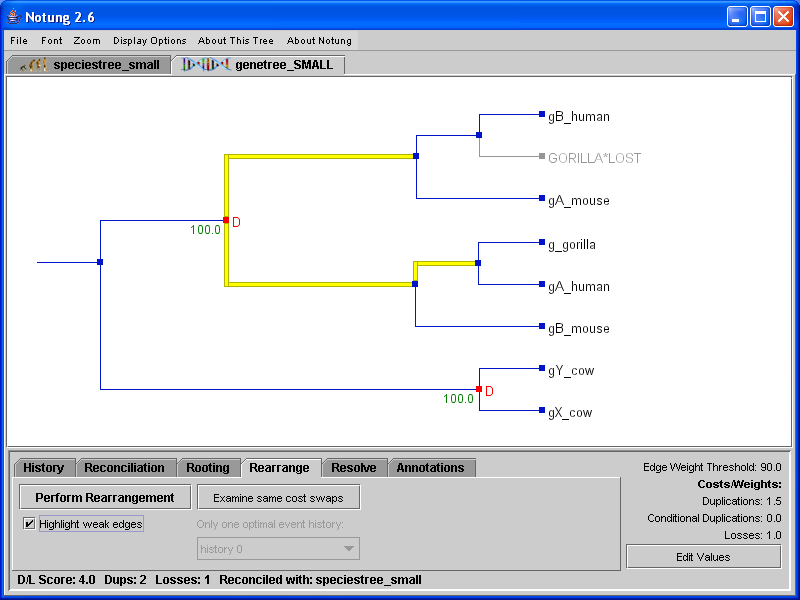
Figure E.5: The gene tree should now look like this.
Change the parameter values and rearrange again
In the previous steps, we rearranged the tree using the default parameter values (cD=1.5 and cL=1.0). For the default values, there is only one minimum cost tree. We now explore what happens when we rearrange the tree when duplications and losses are weighted equally.
A message appears to warn us that although we have changed the parameter values, this has had no effect on the tree. We must rearrange the tree again to see the effect of rearrangement with this choice of parameter values. Click “OK.”
Duplications and losses are now weighted equally in Notung’s reconciliation algorithm.
Click on image to see larger version
Figure E.6: The gene tree should now look like this.
View a different alternate event history
With the new parameter values, there is more than one alternate gene tree with minimal D/L Score. You are currently viewing history 0.
This opens a list of available alternate event histories. You should see history 0 and history 1.
A different tree appears. This tree also has a D/L Score of 3.0, but has two duplications and one loss instead of three duplications and no losses.
Swap nodes in the rearranged tree
Note that this tree groups gB_human with gA_mouse and gA_human with gB_mouse. However, the tree that groups gA_human with gA_mouse and gB_human with gB_mouse has the same score.
Nodes that can be interchanged without changing the D/L Score are marked with enlarged light blue boxes.
To select the node, you must click on the enlarged blue box. When you are able to click and select a node, a blue triangle will mark the node(s). Once selected, the node is marked with a light blue triangle. Each node it can be swapped with is marked with a pink triangle. In this case, there is just one: gA_human.
The nodes gB_human and gA_human are swapped. Once they have been swapped, they are temporarily highlighted with yellow triangles, so that you can see the results of the most recent action. Note that the gA genes are now grouped together, and the gB genes are together in the same subtree, along with the g_gorilla gene.
Click on image to see larger version
Figure E.7: The gene tree should now look like this.
Try performing additional swaps to see how many alternate, minimum cost trees you can find.
In this exercise, you will perform Notung’s main tasks on the gene tree exercise4_genetree with the non-binary species tree exercise4_speciestree. You will reconcile and root the gene tree, and use Notung to determine the upper and lower bounds on the time when a duplication occurred.
Open the tree files
This is an artificial tree made up for this exercise.
As you will notice, this is a non-binary species tree with a polytomy representing the common ancestor of the marsupials.
Reconcile the gene tree with the species tree
The reconciled tree appears in the tree panel. Note that it has a D/L Score of 8.0, with two duplications, one conditional duplication, and five losses. Two red D’s in the tree mark the required duplications, while the one pink cD marks the conditional duplication. At the leaves of the tree, five loss nodes appear in light gray type.
Click on image to see larger version
Figure E.8: The gene tree should now look like this.
Check the duplication bounds
The duplication bounds provide information regarding when gene duplications occurred in the course of species evolution.
In the new window, required duplications are described first. Conditional duplications are described below the required duplications. For both types of duplications, the duplication nodes are listed in the left column, expressed as node names in the gene tree. The lower and upper bounds are listed in the middle and right columns, respectively, and are expressed as internal node names in the species tree. Information on losses is provided below the conditional duplication bounds.
Run the Rooting Analysis
The edge leading to genes from placental mammals (cow, mouse, and human) is colored red. This means it has the lowest root score.
Notice that the red edge has a root score of 7.0. The next lowest root score is 8.0.
The name of the species to which the node is mapped appears in italics next to each internal node.
The tree is rooted on the edge which splits the tree between placental mammals (Eutheria) and marsupials (Metatheria). The D/L Score of the tree is now 7.0, with two duplications, one conditional duplication, and four losses.
Do not close these trees yet - they will be used in upcoming steps.
Click on image to see larger version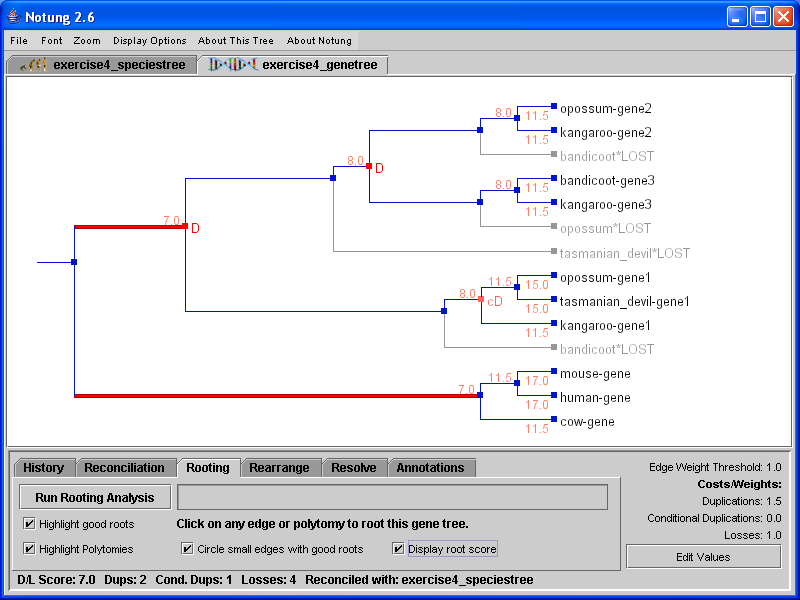
Figure E.9: The gene tree should now look like this.
Reconcile the Tree using the Combined Polytomy Losses algorithm
This step uses the command line interface and can be skipped, if desired. You will use the command line interface to reconcile the gene tree exercise4_genetree with the species tree exercise4_speciestree using the combined losses algorithm.
For instructions on using Notung from the command line, see Chapter 12.2 - Running Notung from the command line.
java -jar Notung-2.6.jar
sampleTrees/exercise4_genetree
-s
sampleTrees/exercise4_speciestree --reconcile
--exact-losses
--outputdir sampleTrees
--report-heuristic-losses
Notung will print information to the screen as it reconciles the tree for both combined and explicit losses. Notice that the first unrooted gene tree has a D/L Score of 8.0, with two duplications, one conditional duplication and five heuristic losses as compared to the second unrooted gene tree, which has a D/L Score of 7.0, with two duplications, one conditional duplication, and four exact losses. The tree, reconciled and with exact losses, will be saved to the sampleTrees folder (as specified by --outputdir) as exercise4_genetree.reconciled.
Root the tree reconciled with the Combined Polytomy Losses algorithm
In the previous step, you reconciled the gene tree while using the combined polytomy losses algorithm. In this step you are will find the optimal root for this gene tree. If you skipped the previous step, you will need to use the gene tree exercise4_genetree-exactLosses.ntg instead of exercise4_genetree.reconciled.
If you skipped the last step, use exercise4_genetree-exactLosses.ntg instead.
A warning will appear stating that the tree was reconciled using --exact-losses. Click the “OK” button.
The tree is rooted on the edge leading to placental mammals. The D/L Score of the tree is now 6.0, with two duplications, one conditional duplication, and three losses.
Click on image to see larger version
Figure E.10: The gene tree should now look like this.
Compare this tree with the previously rooted gene tree (exercise4_genetree). Can you find the difference between the trees? In exercise4_genetree, the loss node, tasmanian_devil*LOST, above the subtree containing genes gene3 and gene2, has been moved below the duplication node and combined with opossum*LOST and bandicoot*LOST in the gene3 and gene2 subtrees, respectively, in exercise4_genetree.reconciled. This resulted in a reduction of the total number of losses.
View polytomy losses without species names included
There are two display options for polytomy losses. In this step, you will see the other way to display these losses.
Click on image to see larger version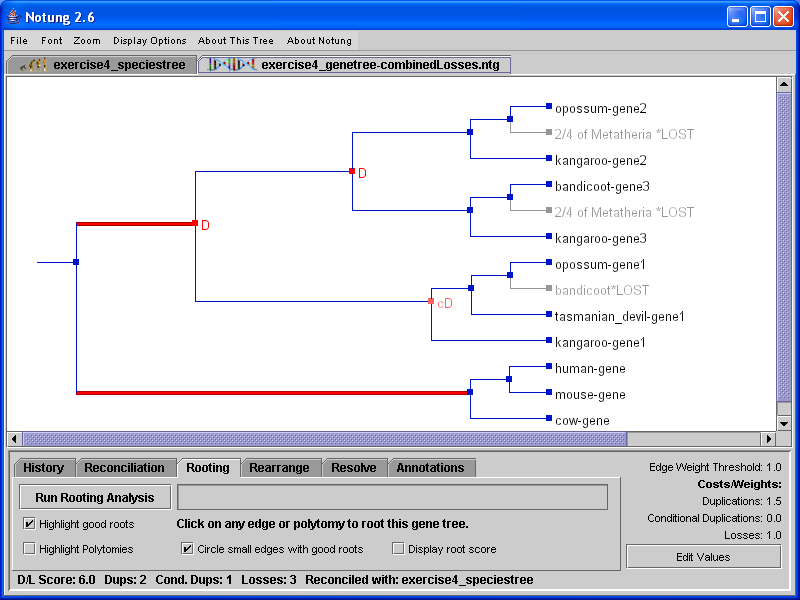
Figure E.11: The gene tree should now look like this.
In this exercise, you will perform Notung’s main tasks on the non-binary gene tree exercise5_genetree with the species tree exercise5_speciestree. You will reconcile, root, resolve, and rearrange the gene tree, and use Notung to determine some general statistics about the trees.
Open the tree files
This is an artificial tree made up for this exercise. Notice that this gene tree is non-binary and contains multiple polytomies.
The polytomies in the gene tree are circled and highlighted in cyan.
Click on image to see larger version
Figure E.12: The gene tree with polytomies highlighted.
Reconcile the gene tree with the species tree
The reconciled tree appears in the tree panel. Note that it has a D/L Score of 20.0, with ten duplications and five losses. Also note that some of the polytomies have more than one duplication associated with the node (ex: the polytomy with eight children has two duplications).
Click on image to see larger version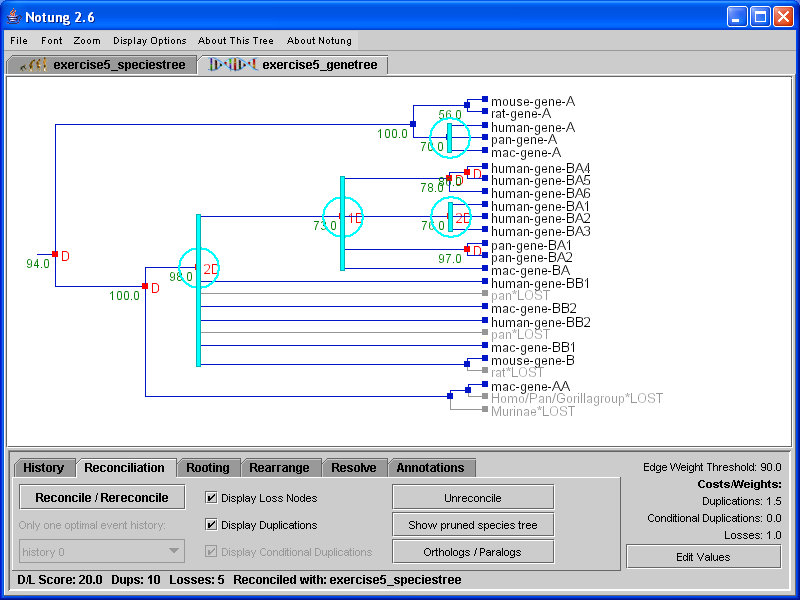
Figure E.13: The gene tree should now look like this.
Get general tree statistics for the gene tree
In this step you will gather some general statistics about the reconciled gene tree and the species tree.
The General Tree Statistics window appears. In this window is information on both the gene tree, the reconciled gene tree, and the species tree. You may have to scroll down to view all the information.
The General Tree Statistics Window should look like this.
Click on image to see larger version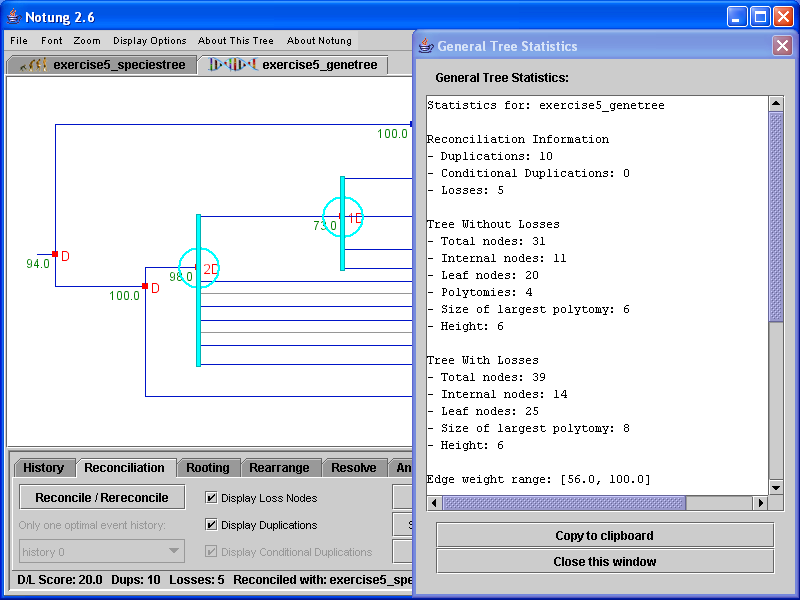
Figure E.14: The General Tree Statistics Window should look like this.
For more information on the data in the General Tree Statistics window, see Chapter 3.4 - General Tree Statistics.
Resolve the polytomies in the gene tree
In this step, you will resolve all the polytomies in the gene tree, thus creating a binary gene tree.
The Resolve task panel opens below.
The polytomies in the gene tree are circled and highlighted in cyan.
The resolved tree appears in the tree panel. Edges associated with the resolved polytomies are now colored cyan. This is the same tree as before, only now the polytomies have been resolved. The number of duplications and losses are identical to the reconciled tree, and even the duplication bounds are the same.
Click on image to see larger version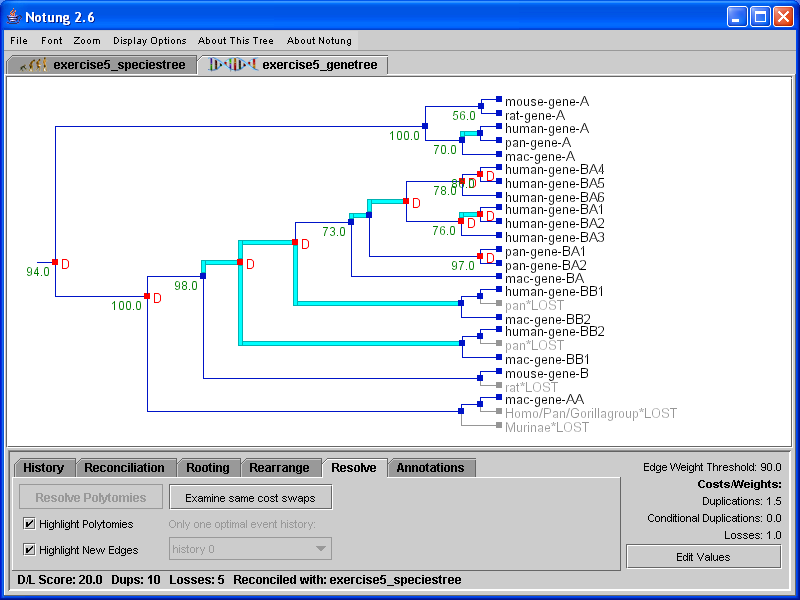
Figure E.15: The gene tree should now look like this.
Change the parameter values and view alternate event histories
In the previous steps, we reconciled and resolved the tree using the default parameter values (CD=1.5 and CL=1.0). For the default values, there is only one minimum cost tree. We now explore what happens when we reconcile the tree when duplications and losses are weighted equally.
We must go back in the history before we change parameter values, as the tree has already been resolved and the change in values might effect the current resolution of the tree.
The tree panel shows the state of the tree before the polytomies were resolved.
Duplications and losses are now weighted equally, and the gene tree is automatically rereconciled with the new parameter values.
The reconciled tree appears in the tree panel. There is now more than one alternate gene tree with the minimal D/L Score. You are currently viewing history 0.
A different tree appears. This tree has a D/L Score of 15.0, with ten duplications and five losses. This tree has the same duplications and losses as the tree reconciled with a duplication cost of 1.5 and a loss cost of 1.0 (see Figure E.14).
A different tree appears. This tree also has a D/L Score of 15.0, but has eleven duplications and four losses rather than the ten duplications and five losses in history 1. The large polytomy with seven children now has three duplications and one loss, whereas in history 1 it had two duplications and two losses.
Click on image to see larger version
Figure E.16: The gene tree should now look like this.
Run the Rooting Analysis
Many edges and one polytomy are colored red, which indicates that all of these components of the tree have the lowest root score.
Notice that the large polytomy is circled in red. Placing a root at a polytomy indicates that at least one edge in the binary resolution of the polytomy has the lowest root score.
Click on image to see larger version
Figure E.17: The gene tree should now look like this.
Each edge and polytomy is labeled with its root score.
The tree is rooted on the polytomy and the D/L Score of the tree is still 15.0, with eleven duplications and four losses.
Click on image to see larger version
Figure E.18: The gene tree should now look like this.
Resolve the polytomies in the gene tree
In this step, you will resolve all the polytomies in the gene tree, thus creating a binary gene tree.
The Resolve task panel opens below.
The polytomies in the gene tree are circled and highlighted in cyan.
The resolved tree appears in the tree panel. Edges associated with the resolved polytomies are now colored cyan.
Click on image to see larger version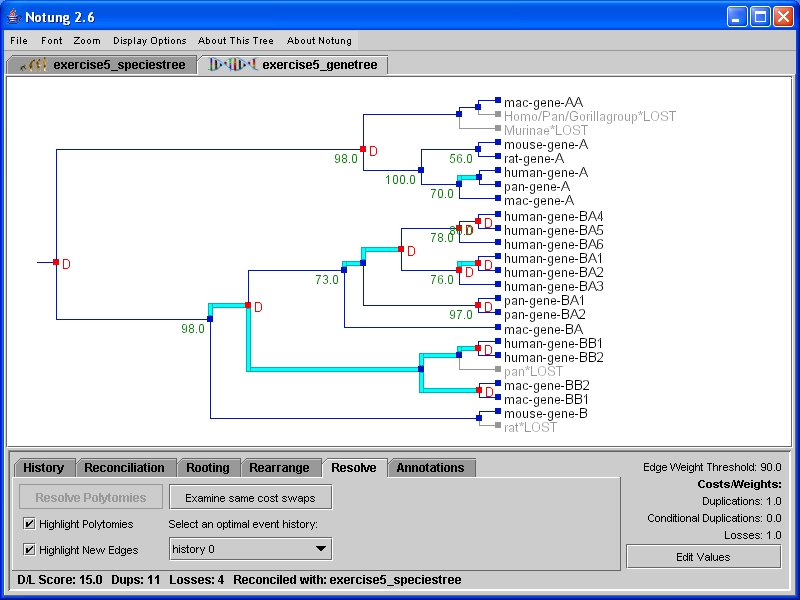
Figure E.19: The gene tree should now look like this.
View a different alternate event history
With these parameter values, there is more than one alternate gene tree with minimal D/L Score. You are currently viewing history 0.
This displays a list of available alternate event histories. You should see history 0 and history 1.
A different tree appears. This tree also has a D/L Score of 15.0, but has ten duplications and five losses instead of eleven duplications and four losses.
Note that these alternate histories correspond to the same alternate histories that were presented after reconciliation.
Swap nodes in the resolved tree
Note that this tree groups human-gene-BB1 with mac-gene-BB2 and human-gene-BB2 with mac-geneBB1. However, the tree that groups human-gene-BB1 with mac-geneBB1 and human-gene-BB2 with mac-gene-BB2 has the same score.
Nodes that can be interchanged without changing the D/L Score or history implied by the polytomies are marked with enlarged light blue boxes.
The node is now marked with a light blue triangle. Each node it can be swapped with is marked with a pink triangle. In this case, there is just one: the node leading to mac-gene-BB2.
The nodes mac-gene-BB1 and mac-gene-BB2 are swapped. Once they have been swapped, they are temporarily highlighted with yellow triangles, so that you can see the results of the most recent action. Note that the BB1 genes are now grouped together, and the BB2 genes are together in the same subtree.
Click on image to see larger version
Figure E.20: The gene tree should now look like this.
Annotate the Gene Tree
This step will introduce you to Notung’s annotations capabilities.
The Annotations task panel is displayed.
A box will appear to edit the new annotation.
This will automatically annotate all the leaves that contain the string “-A” with the color you selected.
This will automatically annotate all the leaves that contain the string “-BA” with the color you selected.
This will automatically annotate all the leaves that contain the string “BB1” with the color you selected.
This will automatically annotate all the leaves that contain the string “BB2” with the color you selected.
This option lets you select the nodes to add to the annotation without searching for a substring.
Notice that these leaves were previously in the color selected in step 3. The leaves are a new color now because the newer annotation takes precedence.
Click on image to see larger version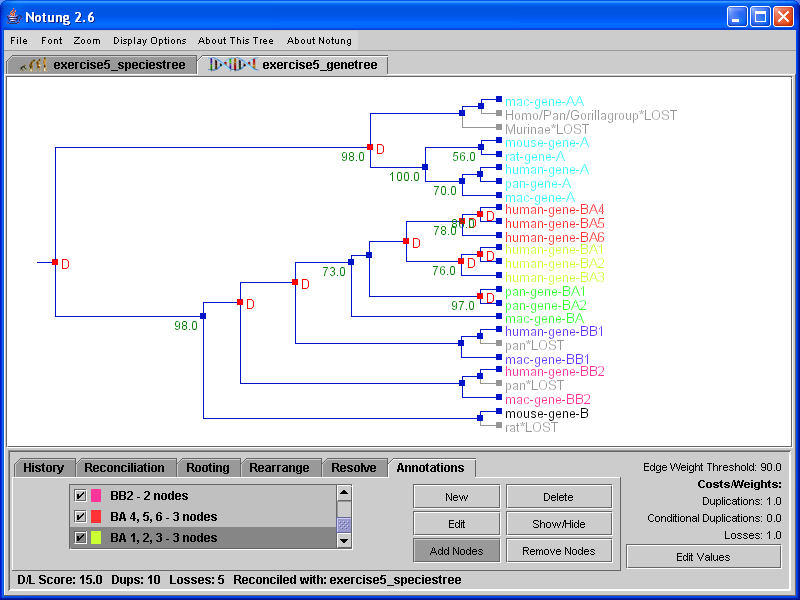
Figure E.21: The gene tree should now look something like this.
Rearrange the resolved tree
In this step, you will rearrange the gene tree to obtain the minimal D/L Score. In this exercise, you have resolved the polytomies in the gene tree before rearranging the weak areas of the tree. However, it is possible to do both task at the same time while in the rearrangement mode. Both Resolve and Rearrangement are available because these two functions have different purposes. If you want to obtain a hypothesis of the binary gene tree, but wish to retain all the information in the gene tree, use the Resolve task mode. However, if you wish to consider edges with an edge weight below a certain value as uninformative, use the Rearrangement task mode.
Several edges in the reconciled tree are highlighted in yellow. These are edges with weights below the Edge Weight Threshold and are considered “weak.“” Weak edges may be rearranged to reduce the number of duplications and losses in the tree. Edges with weights above the threshold will not be rearranged.
Click on image to see larger version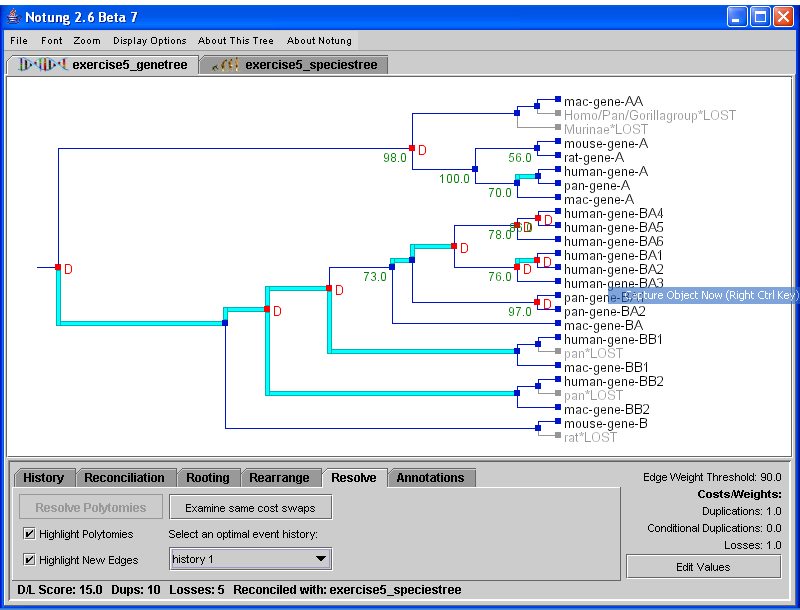
Figure E.22: The gene tree with weak edges highlighted.
The rearranged tree appears in the tree panel. It has a D/L Score of 15.0, with twelve duplications and only three losses. Note that the score did not change; the rearranged tree is not necessarily “better” than the original tree.
Click on image to see larger version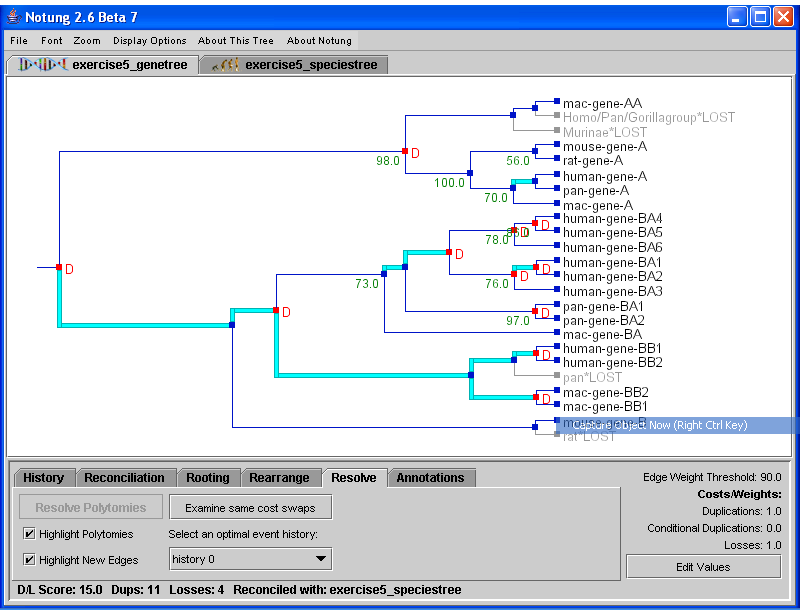
Figure E.23: The gene tree should now look like this.
View a different alternate event history
You are currently viewing history 0.
This opens a list of available alternate event histories. You should see history 0, history 1, and history 2.
Nodes that can be interchanged without changing the D/L Score are marked with enlarged light blue boxes. Try performing additional swaps to see how many alternate, minimum cost trees you can find.
HINT 1: Select the history with ten duplications and five losses.
HINT 2: Swap the subtree of BA1 and BA2 genes in “pan” with the LOST “pan” gene in the BA subtree.
HINT 3: Swap the subtree of BA4, BA5, and BA6 in human with the node for BA3 in human.
Appendix G Notung as an Applet
In addition to a stand-alone application, Notung is available as a Java applet that can be embedded in an HTML page and executed in any java-enabled web browser. You can use the Notung applet to present phylogenetic data on the web, by creating a webpage that allows visitors to your site to view, analyze or manipulate trees interactively using Notung.
Section G.1 is intended for Notung applet users. It describes the Notung applet functions and user interface, focusing primarily on differences between the applet and the standalone application. Section Section G.2 is targeted at web site developers and describes how to embed the Notung applet in an HTML file.
Problem Possible Causes Solutions When I tried to reconcile the trees, I received this error message: “None of the species labels in this tree can be found in the species tree. Try checking your reconciliation settings.”
- The species labels in the gene tree leaf node names are not compatible with the species labels in the species tree.
- The “Specify Species Label” setting in the Reconciliation Options dialog box has been set incorrectly.
- The incorrect species tree has been selected for reconciliation.
- Check the species labels in the gene tree to make sure they match the species labels in the species tree.
- In the Reconciliation Options dialog, make sure you select the appropriate naming convention for species labels.
- In the Reconciliation Options dialog, make sure you select the appropriate species tree for reconciliation.
The edge weights on the gene tree are not what you expected. Notung has mistaken the branch length values in the Newick file for edge weight values. See Appendix A.6 - Location of Edge Weight Values First, open the gene tree file in a text editor to determine the location of edge weight values. Then, click “Display Options → Select Location of Edge Weights” and set the location of Edge Weights appropriately. My gene tree should have edge weights, but when I load the tree, weights are not displayed on some branches. The gene tree file is supposed to be in Newick, NHX or Notung format, but contains a typo or formatting error, affecting the edge weight location. Open the original tree file in a tree editing program or text editor and correct any formatting errors. NOTE: Some formats are case-sensitive. When I tried to reconcile the gene tree with the species tree, I received this message: “There are no species trees to reconcile with.” -or- My species tree is not listed in the drop down menu in the Reconciliation Options dialog box. You have opened a species tree as a gene tree. Reopen the desired species tree as a species tree using “File → Open Species Tree” or “Ctrl-Shift-O”. After reconciliation, I found lost genes in unrecognizable species, such as “n101.” The gene was lost in an ancestral species that was not given a label in the original species tree file. When internal node names are not specified in the input file, Notung generates them using an arbitrary counting system (ex: n101). Use “Display Options → Display Internal Node Names” to examine internal species names in the species tree. If you prefer taxonomic names, use a tree editing program or text editor to add real species names to internal nodes in the species tree. When I tried to open a tree, I received this message: “An error occurred while opening your file. Please check the format.” Or “An error occurred while opening your file. Node had malformed information.”
- The gene tree file is supposed to be in Newick, NHX or Notung format, but contains a typo or formatting error.
- The gene tree file is in a format Notung does not accept, (ex: Nexus).
- Open the original tree file in a tree editing program or text editor and correct any formatting errors.
- Convert the file to Newick or NXH file format. See Appendix A - File Formats for more information about file formats.
Notung reports that you do not have a recent enough version of Java, but you have the latest version installed. You have multiple versions of Java installed.
- On Windows, bring up the properties window for the Notung-2.6 jar file. Check the “Opens With” field - if the wrong version of java is listed, change it so that the right version of java is being used.
- On Linux, type java -version - this will tell you which version of Java is being used. If it is incorrect, alter your path environment variable to include the proper version of Java.
The species tree file I created using the NCBI Taxonomy Browser contained non-ASCII characters. As part of its file construction, the NCBI Taxonomy Browser includes some non-ASCII characters. These characters are ignored by Notung, but you can open the tree file in a text editor and delete the non-ASCII characters. The species tree file I created using the NCBI Taxonomy Browser contained 4’s. As part of its file construction, the NCBI Taxonomy Browser includes a branch length of 4 for every edge in the species trees it produces. These branch lengths are ignored by Notung, but you can open the tree file in a tree editing program or text editor and delete the branch lengths. The names of internal nodes in my gene tree change over time.
- The gene tree file does not specify internal node names and has been reloaded. When internal node names are not specified in the input file, Notung generates them using an arbitrary counting system (ex: n101).
- Node names were given in the original tree file, but additional nodes have been added, because either rearrangement or resolve has been performed. Added nodes are assigned names that begin with an ‘r’ and are followed by numbers (ex: r245).
- If you want the internal node names to be the same every time the tree is opened, use a tree editing program or text editor to add names to internal nodes in the gene tree.
- Notung cannot track internal nodes that are temporary or not present in the original file. If you need permanent names for these nodes, save the file and use a tree editing program or text editor to specify names for these nodes.
I use the <Tab> key to navigate to a different button in a popup box, but when I hit the <Enter> key, the selected button is not engaged. This is a problem with some versions of Java. The <Tab> key option to navigate to different buttons does not select the “highlighted” button. When the <Enter> key is pressed, the originally selected button is used. Use the mouse to select buttons in the windows I have added a node to an annotation, but the node does not appear in the correct color. There are conflicting annotations - the node corresponds to more than one annotation and is currently being described by another annotation. Annotations have precedence - those annotations added later will always take precedence over earlier annotations. Manually remove the node from the other annotations, or check the other annotations and remove any search strings that identify the node of interest. See Chapter 10 - Annotations for more information.
Because Java applets have limitations that stand-alone applications do not have, there are differences between using the two.
In this section, we describe how to construct a web page with an embedded Notung applet. The following files are required:
The HTML required to embed the Notung applet in a web page must include a definition of notung.js. This is typically of the form
<head> <script src='notung.js'></script> </head> <body> <a href="javascript:openNotung( ... )" title="Notung JavaApplet"> Informative title goes here</a> </body>
The main work is carried out by the function openNotung, which takes four parameters:
Here are two examples of HTML code that defines javascript that calls openNotung(), creating a link to launch the Notung applet.
Example 1:
<head> <script src='notung.js'></script> </head> <body> <a href="javascript:openNotung( ['GENE_TREE_001'], ['SPECIES_TREE_001'], '', 'Example Trees One')" title="Notung JavaApplet"> Open GENE_TREE_001 and SPECIES_TREE_001 in Notung</a> </body>
In this example, the web page, jar file, notung.js, and trees are all located in the same directory.
Example 2:
<head> <script src='http://www.yourdomain.com/applet_files/notung.js'></script> </head> <body> <a href="javascript:openNotung( ['http://www.yourdomain.com/tree_files/GENE_TREE_001', 'http://www.yourdomain.com/tree_files/GENE_TREE_002'], [], '', 'Example Trees Two')" title="Notung JavaApplet"> Open GENE_TREE_001 and GENE_TREE_002 in Notung</a> </body>
In this example the Notung jar file and notung.js are both
located in the directory
http://www.yourdomain.com/applet_files.
The gene trees are located in the directory
http://www.yourdomain.com/tree_files. The web page can be
located anywhere on the webserver http://www.yourdomain.com/
This example displays two gene trees, and no species trees.
Because of restrictions on the actions of Java applets, all of the files used for the Notung applet (the jar file, notung.js, and the tree files) must be located on the same webserver as the web page.
<script src=`http://www.yourdomain.com/path/to/files/notung.js'> </script>
// url for jar file var jar = "Notung-2.6.jar"
This document was translated from LATEX by HEVEA.