Computational Method Puts Finer Point on Multispecies Genomic Comparisons Probabilistic Model Could Provide Insights Into What Makes a Human a Human
Byron SpiceWednesday, June 20, 2018Print this page.
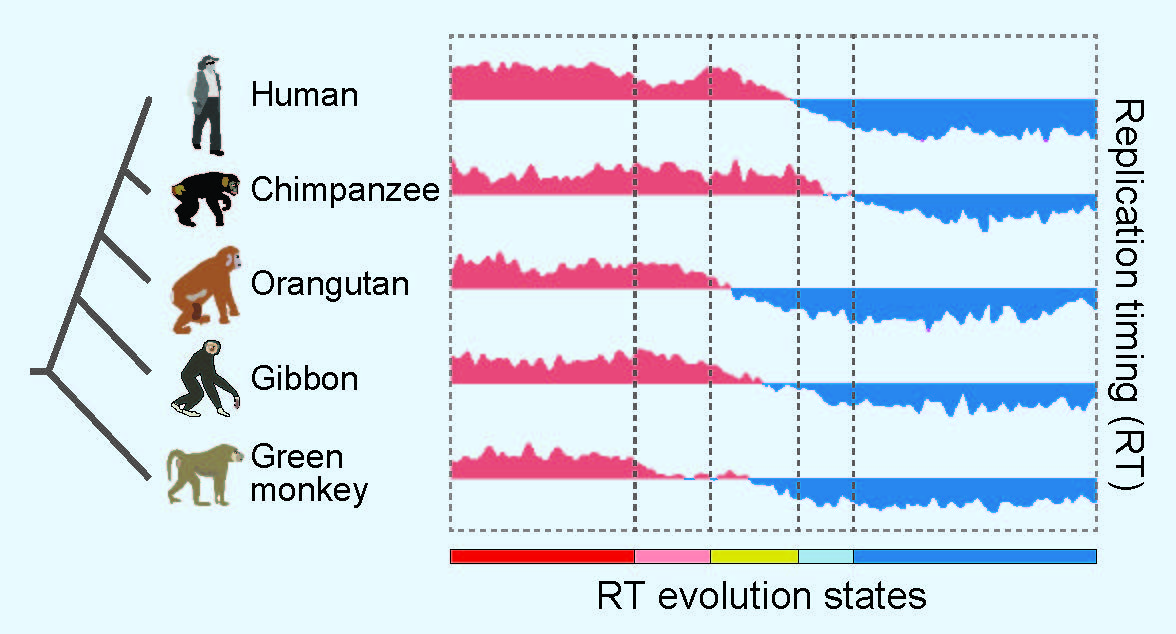
A new computational tool will potentially help geneticists to better understand what makes a human a human, or how to differentiate species in general, by providing more detailed comparative information about genome function.
In a report published online today by the journal Cell Systems, researchers led by Jian Ma, associate professor of computational biology at Carnegie Mellon University, describe a new model for performing comparative analyses of genome function across multiple species. Such analysis may provide insights into not only evolution, but also human disease.
The research team, including scientists from the University of Virginia, Florida State University and the University of Connecticut, developed the Phylogenetic Hidden Markov Gaussian Processes model, or Phylo-HMGP, to analyze functional genomic data. They used the model to analyze a new dataset for DNA replication timing across five primate species, including human.
Genetic differences in protein-coding genes alone cannot account for the dramatic variation between species, so scientists increasingly focus on differences in gene regulation — mechanisms that control how and to what degree genes are activated.
"The differences among primate species may be mostly in the noncoding regions of the genome, the regulatory elements, not the genes themselves," Ma explained. High-throughput technologies produce a large amount of functional genomic data, which should help scientists better understand how genomes evolved.
Ma said Phylo-HMGP addresses what might be called the "Starbucks problem" in these multispecies analyses. Just as coffee vendors tend to sell drinks in small, medium and large sizes, analysis tools typically characterize functional genomic data as low, medium or high.
"With Phylo-HMGP, we can look at each functional genomic value as a continuous signal — showing the actual activity level rather than just a rough level estimate," said Yang Yang, a Ph.D. student in CMU's Computational Biology Department and first author of the study. "In this way, we're able to fully utilize the data that have been gathered."
The researchers applied the model to an analysis of DNA replication timing, the order in which segments of DNA are replicated, which can vary from species to species. They did so for a dataset including humans, chimpanzees, orangutans, gibbons and green monkeys that was generated in collaboration with David M. Gilbert of Florida State University and Rachel J. O'Neill of the University of Connecticut.
"We demonstrated that we could use Phylo-HMGP to discover genomic regions with distinct evolutionary patterns of replication timing," Ma said. Their research provides a framework for applying the model to reveal genomic regions with functions that are similar across species and those that are varied, or dynamic, between species. Analyses of dynamic regions in functional genomic datasets not only can improve understanding of evolution, but also may have implications for certain types of species-specific diseases, he added.
Other research team members include Yang Zhang, a research associate in the Computational Biology Department; Quanquan Gu of the University of Virginia; Takayo Sasaki of Florida State; and Julianna Crivello of the University of Connecticut. The National Institutes of Health and the National Science Foundation supported this research.
A copy of the research paper is available on the Cell Systems website.
Byron Spice | 412-268-9068 | bspice@cs.cmu.edu