Research Areas
Machine LearningEmphasizing theory and
algorithms for learning from high-dimensional data, and reasoning under
uncertainty. |
Computational BiologyEmphasizing
developing formal models and algorithms that address problems of
practical biological and medical concerns. |
|
|
|
|
Machine LearningEmphasizing theory and algorithms for learning complex probabilistic models, learning with prior knowledge, and reasoning under uncertainty. |

Learning Time and Space Varying-Coefficient Models with Evolving Structures, and Applications in Socio-cultural Data Mining and Systems Biology
 In this project we develop
methodologies for estimating
and
analyzing varying-coefficient models with structural changes occurring
at
unknown times or locations. Instances of such models are frequently
encountered
in social and biological problems where data are structured and
longitudinal,
and the iid assumptions on sample with respect to an invariant
underlying model
no longer hold. For example, at each time point, the observation (such
as a
single snapshot of social state of all actors) is distributed according
to a
model (such as a network) specific to that time, and therefore can not
be directly
used for estimating models underlying other time points. The main
issues we
concern in this project include estimating the changing model
structures and
parameters, number of structural changes, the change times, and the
unknown
coefficient functions. We will develop efficient and scalable
algorithms for
addressing these problems under "small n large p" scenarios based on
techniques such as sparse regression under various regularization and
structured-constraint schemes, convex optimization, and Bayesian
inference. We
will also focus on asymptotic analysis of the procedure and give
conditions
under which it is able to correctly estimate the structural changes and
the
model coefficients.
In this project we develop
methodologies for estimating
and
analyzing varying-coefficient models with structural changes occurring
at
unknown times or locations. Instances of such models are frequently
encountered
in social and biological problems where data are structured and
longitudinal,
and the iid assumptions on sample with respect to an invariant
underlying model
no longer hold. For example, at each time point, the observation (such
as a
single snapshot of social state of all actors) is distributed according
to a
model (such as a network) specific to that time, and therefore can not
be directly
used for estimating models underlying other time points. The main
issues we
concern in this project include estimating the changing model
structures and
parameters, number of structural changes, the change times, and the
unknown
coefficient functions. We will develop efficient and scalable
algorithms for
addressing these problems under "small n large p" scenarios based on
techniques such as sparse regression under various regularization and
structured-constraint schemes, convex optimization, and Bayesian
inference. We
will also focus on asymptotic analysis of the procedure and give
conditions
under which it is able to correctly estimate the structural changes and
the
model coefficients.
Selected Reading:
- F.
Guo, S. Hanneke, W. Fu and E.
P. Xing, Recovering
Temporally
Rewiring
Networks: A model-based approach,
Proceedings of the 24th International Conference on Machine Learning (ICML
2007)
- M.
Kolar, L. Song, A. Ahmed, and E. P. Xing, Estimating
Time-Varying Networks,
Manuscript, arXiv:0812.5087.
[Top]
Learning Sparse Structured Input/Output Models in Very High-dimensional Space, and Applications in Structured prediction problems in NLP, Vision, and Bioinformatics
 In many
high-dimensional structured I/O
problems, such as
genome-phenome association analysis and image segmentation, where both
input
and output can contain tens of thousands, sometimes even millions of
inter-related
features, learning a sparse and consistent structured predictive
function can
be of paramount importance for both robustness and interpretability of
the
model. In this project, we attack this problem from two directions. One
direction we will pursue to learn sparse structured I/O models is to
extend the
l1-regularized regression model (i.e., the lasso method) to a family of
sparse
"structured" regression models in the contexts of uncovering true
associations between linked genetic variations (inputs) and networked
phenotypes (outputs), which can be cast as efficiently solvable convex
optimization problems and yield parsimonious and possibly consistent
maximum
likelihood estimates of the model. Another direction we are exploring
is based
on a new statistical formalism known as the maximum entropy
discrimination
Markov networks, which address the problem of estimating sparse
structured I/O
models under a maximum margin framework, but using a entropic
regularizer that
leads to a distribution of structured prediction functions that are
simultaneously primal and dual sparse (i.e., with few support vectors,
and of
low effective feature dimension), and can be efficiently solved via a
novel
algorithm that builds on variational inference and existing solvers for
the
maximum margin Markov network (which is a special case of our proposed
model).
We will also investigate the theoretical guarantee of these methods
such as
generalization bounds, sample complexity, convergence behavior.
In many
high-dimensional structured I/O
problems, such as
genome-phenome association analysis and image segmentation, where both
input
and output can contain tens of thousands, sometimes even millions of
inter-related
features, learning a sparse and consistent structured predictive
function can
be of paramount importance for both robustness and interpretability of
the
model. In this project, we attack this problem from two directions. One
direction we will pursue to learn sparse structured I/O models is to
extend the
l1-regularized regression model (i.e., the lasso method) to a family of
sparse
"structured" regression models in the contexts of uncovering true
associations between linked genetic variations (inputs) and networked
phenotypes (outputs), which can be cast as efficiently solvable convex
optimization problems and yield parsimonious and possibly consistent
maximum
likelihood estimates of the model. Another direction we are exploring
is based
on a new statistical formalism known as the maximum entropy
discrimination
Markov networks, which address the problem of estimating sparse
structured I/O
models under a maximum margin framework, but using a entropic
regularizer that
leads to a distribution of structured prediction functions that are
simultaneously primal and dual sparse (i.e., with few support vectors,
and of
low effective feature dimension), and can be efficiently solved via a
novel
algorithm that builds on variational inference and existing solvers for
the
maximum margin Markov network (which is a special case of our proposed
model).
We will also investigate the theoretical guarantee of these methods
such as
generalization bounds, sample complexity, convergence behavior.
Selected Reading
- S.
Kim, K. Sohn, and E. P. Xing, A Multivariate
Regression Approach to Association Analysis of Quantitative Trait
Network ,
Manuscript, arXiv:0811.2026.
- J.
Zhu and E. P. Xing, Maximum Entropy Discrimination
Markov Networks,
Manuscript, arXiv:0901.2730.
- J.
Zhu, E. P. Xing, B. Zhang,
Laplace
Maximum Margin Markov Networks ,
Proceedings of the 25th International Conference on Machine Learning (ICML
2008).
[Top]
Statistical Models and Algorithms of Networks and Relational Data (in collaboration with Stephen Fienberg)
 A plausible representation of the relational
information among
entities in dynamic systems such as a living cell, or a social
community, or
the internet, is a stochastic network which is topologically rewiring
and
semantically evolving over time (or space). In this project we develop
probabilistic generative models for the formation, growth, evolution,
and
dynamics of networks and relational data in general, and
inference/learning
algorithms for node labeling, link prediction, latent theme extraction,
etc.,
for network and relational data. We also work on theoretical issues,
such as
bounds, complexity, related to our models and algorithms, and
applications to
the analysis of socio-cultural networks, author-citation networks, the
blogosphere, and biological networks.
A plausible representation of the relational
information among
entities in dynamic systems such as a living cell, or a social
community, or
the internet, is a stochastic network which is topologically rewiring
and
semantically evolving over time (or space). In this project we develop
probabilistic generative models for the formation, growth, evolution,
and
dynamics of networks and relational data in general, and
inference/learning
algorithms for node labeling, link prediction, latent theme extraction,
etc.,
for network and relational data. We also work on theoretical issues,
such as
bounds, complexity, related to our models and algorithms, and
applications to
the analysis of socio-cultural networks, author-citation networks, the
blogosphere, and biological networks.
Selected Reading
- E.
Airoldi, D. Blei, S. Fienberg, and E. P. Xing, Mixed
Membership Stochastic Blockmodel, Journal of Machine
Learning Research,
9(Sep):1981--2014, 2008.
- W.
Fu, L. Song, and E. P. Xing, A State-Space Mixed Membership
Blockmodel for Dynamic Network Tomography,
Manuscript, arXiv:0901.0135.
[Top]
Bayesian statistics, nonparametric Bayesian analysis, algorithms and applications of Bayesian nonparametrics in data mining
 In this project we develop nonparametric and
semiparametric Bayesian models (based on the Dirichlet process and
extensions,
sometimes known as the generalized Polya urn schemes) for analyzing
time series data,
hierarchical data, and other complex inputs with uncertain internal
structure,
which arise from temporal text mining (e.g., emails, news streams),
object
tracking (e.g., video surveillance, navigation and control) and
biological data
analysis. We develop formal probabilistic formalisms, sampling and
variational
inference algorithms, and also address theoretical issues such as
consistence,
bounds and convergence of our models and algorithms.
In this project we develop nonparametric and
semiparametric Bayesian models (based on the Dirichlet process and
extensions,
sometimes known as the generalized Polya urn schemes) for analyzing
time series data,
hierarchical data, and other complex inputs with uncertain internal
structure,
which arise from temporal text mining (e.g., emails, news streams),
object
tracking (e.g., video surveillance, navigation and control) and
biological data
analysis. We develop formal probabilistic formalisms, sampling and
variational
inference algorithms, and also address theoretical issues such as
consistence,
bounds and convergence of our models and algorithms.
Selected Reading
- K.
Sohn and E. P. Xing,
A Hierarchical Dirichlet Process Mixture Model For Haplotype
Reconstruction
From Multi-Population Data,
Annals of Applied Statistics, 2009.
- A. Ahmed
and E. P. Xing, Dynamic
Non-Parametric Mixture Models and the Recurrent Chinese
Restaurant Process, Proceedings
of The Eighth SIAM International Conference on Data Mining (SDM2008).
- E.
P. Xing and K. Sohn, Hidden
Markov Dirichlet
Process: Modeling Genetic Recombination in
Open Ancestral Space,
Bayesian Analysis, Volunn 2, Number 2, 2007.
[Top]
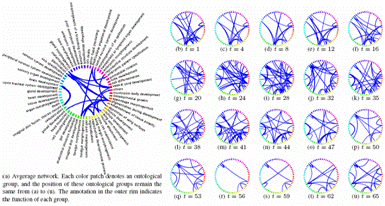
Dynamic topic models for structured browsing of large corpus of text/image/video/network data
 This is a "google-style" project in which we develop
formal methods for visualizing, categorizing, and tracking content-rich
multimedia and network data-streams in an arbitrary-dimension
"tomographic"
space of latent semantic topics, where each entity can be represented
by a human-perceivable and interpretable "semantic" coordinates or
trajectory, so that one can succinctly browse the complex multimodal
and time-evolving data in a direct, global and on-line fashion. Such
display of complex data complements the current Google representation
of
web-information, which is one-dimensional (in terms of a rank list),
static (no change
in the topical content of the media can be traced), and unimodal (only
one
type of media can be displayed as a time, such as text only, or image
only).
One can use our system to directly visualize in the topic space a much
larger
amount of media or web information, rather than via pages of
ordered-lists of
subjects as in current Google interface. One can detect bias,
ideological
perspective, or other subtle information, which hide beneath the
topical contents in
news article or other media. One can directly measure (by eye) the
distance
between query and related entities rather than following a rank list,
which
offer more flexibility and higher accuracy. One can also track the
trajectories of
evolving entities and detection events such as birth/death of
themes/topics
using our system, which is not possible under Google.
This is a "google-style" project in which we develop
formal methods for visualizing, categorizing, and tracking content-rich
multimedia and network data-streams in an arbitrary-dimension
"tomographic"
space of latent semantic topics, where each entity can be represented
by a human-perceivable and interpretable "semantic" coordinates or
trajectory, so that one can succinctly browse the complex multimodal
and time-evolving data in a direct, global and on-line fashion. Such
display of complex data complements the current Google representation
of
web-information, which is one-dimensional (in terms of a rank list),
static (no change
in the topical content of the media can be traced), and unimodal (only
one
type of media can be displayed as a time, such as text only, or image
only).
One can use our system to directly visualize in the topic space a much
larger
amount of media or web information, rather than via pages of
ordered-lists of
subjects as in current Google interface. One can detect bias,
ideological
perspective, or other subtle information, which hide beneath the
topical contents in
news article or other media. One can directly measure (by eye) the
distance
between query and related entities rather than following a rank list,
which
offer more flexibility and higher accuracy. One can also track the
trajectories of
evolving entities and detection events such as birth/death of
themes/topics
using our system, which is not possible under Google.
Selected Reading
- A. Ahmed
and E. P. Xing, Dynamic
Non-Parametric Mixture Models and the Recurrent Chinese
Restaurant Process, Proceedings
of The Eighth SIAM International Conference on Data Mining (SDM2008).
- W.
Fu, L. Song, and E. P. Xing, A State-Space Mixed Membership
Blockmodel for Dynamic Network Tomography,
Manuscript, arXiv:0901.0135.
[Top]
Other applications of probabilistic graphical models in Computational Biology, IR, NLP, Multimedia and Control (in collaboration with many faculty and students at CMU and other universities)
 We design various task-specific generative, discriminative,
and hybrid graphical models and algorithms for various biological and
genetic problems (see bellow), for NLP problems such as statistical
machine
translation, for comprehending and categorizing text corpus, for
segmenting, tracking and interpreting video and caption streams from
various
sources (e.g., surveillance system, robots), and for decision making
and active
learning in dynamic environments.
We design various task-specific generative, discriminative,
and hybrid graphical models and algorithms for various biological and
genetic problems (see bellow), for NLP problems such as statistical
machine
translation, for comprehending and categorizing text corpus, for
segmenting, tracking and interpreting video and caption streams from
various
sources (e.g., surveillance system, robots), and for decision making
and active
learning in dynamic environments.
Selected Reading
- R.
Nallapati, A. Ahmed, E. P. Xing, and W. Cohen, Sparse
Feature Joint Latent Topic Models for Text and Citations ,
Proceedings of The Fourteen ACM SIGKDD International Conference on
Knowledge Discovery and
Data Mining, (KDD
2008).
- J.
Yang, R. Yan, Y. Liu, and E. P. Xing, Harmonium
Models for Video Classification, Journal of Statistical Analysis and
Data Mining, Vol 1, issue 1, p23-37, 2008.
- B.
Zhao and E.
P. Xing, HM-BiTAM: Bilingual Topic
Exploration, Word Alignment, and Translation, Advances
in Neural Information Processing
Systems 20 (NIPS2007).
- L.
Gu, E. P. Xing, and T. Kanade, Learning
GMRF Structures for
Spatial Priors,
Proceedings of the IEEE Conference on Computer Vision and Pattern
Recognition (CVPR
2007).
[Top]
![]()
|
|
Computational BiologyWith an emphasis on developing formal models and algorithms that address problems of practical biological and medical concerns. |

Genome-Transcription-Phenome-Wide Association: a new paradigm for association studies in complex diseases (in collaboration with various faculty in UPMC and Harvard Medical School)
 Many complex disease
syndromes consist of a large number of
related, rather than independent, clinical phenotypes. Differences
between these syndromes involve the complex interplay of a large number
of
genomic variations that perturb the function of disease-related genes
in the
context of a regulatory network, rather than individually. The current
state-of-the-art in genome-wide-association studies (GWAS) is a
single-gene association
approach. A major challenge for the immediate future is therefore to
transcend the
one-gene/one-disease approach with a suite of approaches that account
for
pathway and network structure. In this project, we aim at developing
algorithms
and software to enable next-generation association analysis addressing
these
problems, in the context of asthma disease. We are developing
methodologies for
the largely unexplored but practically important problem of structured
associations between elements in the genome, transcriptome, and the
phenome.
This research will open a new paradigm for association studies of
complex
diseases, which facilitates: 1) Intra- and inter-omic integration of
data for association mapping and disease gene/pathway discovery, 2)
Thorough
explorations of the internal structures within different omic data, so
that cryptic associations that are not possibly detectable in
unstructured
analysis due to their weak statistical power can be now inferred. 3)
Joint
statistical inference of mechanisms and pathways of how variations in
DNA lead to
variations in complex traits flows through molecular networks, and
inference of condition-specific state of gene function in the molecular
networks,
and 4) Development of faster and automated computational algorithm with
greater scalability and robustness to large-scale inter-omic analysis,
and more
convenient software package and user interface.
Many complex disease
syndromes consist of a large number of
related, rather than independent, clinical phenotypes. Differences
between these syndromes involve the complex interplay of a large number
of
genomic variations that perturb the function of disease-related genes
in the
context of a regulatory network, rather than individually. The current
state-of-the-art in genome-wide-association studies (GWAS) is a
single-gene association
approach. A major challenge for the immediate future is therefore to
transcend the
one-gene/one-disease approach with a suite of approaches that account
for
pathway and network structure. In this project, we aim at developing
algorithms
and software to enable next-generation association analysis addressing
these
problems, in the context of asthma disease. We are developing
methodologies for
the largely unexplored but practically important problem of structured
associations between elements in the genome, transcriptome, and the
phenome.
This research will open a new paradigm for association studies of
complex
diseases, which facilitates: 1) Intra- and inter-omic integration of
data for association mapping and disease gene/pathway discovery, 2)
Thorough
explorations of the internal structures within different omic data, so
that cryptic associations that are not possibly detectable in
unstructured
analysis due to their weak statistical power can be now inferred. 3)
Joint
statistical inference of mechanisms and pathways of how variations in
DNA lead to
variations in complex traits flows through molecular networks, and
inference of condition-specific state of gene function in the molecular
networks,
and 4) Development of faster and automated computational algorithm with
greater scalability and robustness to large-scale inter-omic analysis,
and more
convenient software package and user interface.
Selected Reading
- S.
Kim, K. Sohn, and E. P. Xing, A Multivariate
Regression Approach to Association Analysis of Quantitative Trait
Network ,
Manuscript, arXiv:0811.2026.
- S.
Kim and E. P. Xing, Structured
Feature Selection in
High-Dimensional Space via Block Regularized
Regression,
Proceedings of the 24th International Conference on Conference on
Uncertainty in Artificial Intelligence, (UAI
2008).
[Top]
Recovering and Exploring Time-Varying Gene Interactions in Drosophila Development and Human Disease Progression (in collaboration with various faculty in University of Chicago and Harvard Medical School)
 Due to the dynamic
nature of biological systems, biological
networks underlying temporal process such as development,
immunoresponse, and disease progression can exhibit significant
topological changes to
facilitate dynamic regulatory functions. The latent functionality or
membership
undertaken by the biomolecules as determined by these dynamic
interactions will
also exhibit rich temporal behaviors, assuming a distinct function at
one
point while leaning more towards a second function at an another point.
In
this project, we focus on two dynamic processes, the life cycle of
Drosophila melanogaster, and the progress of asthma in human, for which
we develop
methodologies to, 1) reverse-engineer latent time-evolving gene
networks based
on either microimages of spatial pattern of gene expressions in
Drosophila
embryos, or genome-wide microarray profile of gene expression
intensities; 2)
recover the transcriptional activation/repression functions from
temporal/spatial patterns of gene expressions; 3) estimate embeddings
of every genes into a latent function space and track its mixed
membership of
functions in the latent space across time. The goal is to understand
the driving
forces underlying dynamic rewiring of gene regulation circuity, and to
predict
future network structures.
Due to the dynamic
nature of biological systems, biological
networks underlying temporal process such as development,
immunoresponse, and disease progression can exhibit significant
topological changes to
facilitate dynamic regulatory functions. The latent functionality or
membership
undertaken by the biomolecules as determined by these dynamic
interactions will
also exhibit rich temporal behaviors, assuming a distinct function at
one
point while leaning more towards a second function at an another point.
In
this project, we focus on two dynamic processes, the life cycle of
Drosophila melanogaster, and the progress of asthma in human, for which
we develop
methodologies to, 1) reverse-engineer latent time-evolving gene
networks based
on either microimages of spatial pattern of gene expressions in
Drosophila
embryos, or genome-wide microarray profile of gene expression
intensities; 2)
recover the transcriptional activation/repression functions from
temporal/spatial patterns of gene expressions; 3) estimate embeddings
of every genes into a latent function space and track its mixed
membership of
functions in the latent space across time. The goal is to understand
the driving
forces underlying dynamic rewiring of gene regulation circuity, and to
predict
future network structures.
Selected Reading
- A.
Ahmed, L. Song, and E. P. Xing, Time-Varying Networks:
Recovering Temporally Rewiring Genetic Networks During the Life Cycle
of Drosophila melanogaster,
Manuscript, arXiv:0901.0138.
- W.
Fu, L. Song, and E. P. Xing, A State-Space Mixed Membership
Blockmodel for Dynamic Network Tomography,
Manuscript, arXiv:0901.0135.
[Top]
Computational systems biology of genome-microenvironment interactions in breast cancer (in collaboration with Mina Bissell)
 In this project we
analyze the molecular abundance profiles
(e.g., microarray, CGH, ChIp-Chip) measured in a "designer
microenvironment", realized in 3D culture model that imitates the in
vivo cellular context and dynamics of cancer progression, reversion and
apoptosis. We will develop algorithms to identify molecular
determinants and
markers of cancer states and categorize cancers on the basis of
signaling pathway
characteristics. Using probabilistic graphical modeling approaches, we
hope to infer stochastic network models for transcriptional regulation
in
response to combinations of signaling inhibitions in cancer cells.
In this project we
analyze the molecular abundance profiles
(e.g., microarray, CGH, ChIp-Chip) measured in a "designer
microenvironment", realized in 3D culture model that imitates the in
vivo cellular context and dynamics of cancer progression, reversion and
apoptosis. We will develop algorithms to identify molecular
determinants and
markers of cancer states and categorize cancers on the basis of
signaling pathway
characteristics. Using probabilistic graphical modeling approaches, we
hope to infer stochastic network models for transcriptional regulation
in
response to combinations of signaling inhibitions in cancer cells.
Selected Reading:
- TBA
[Top]
Probabilistic evolutionary models of cis-regulatory modules in Drosophila (in collaboration with Martin Kreitman)
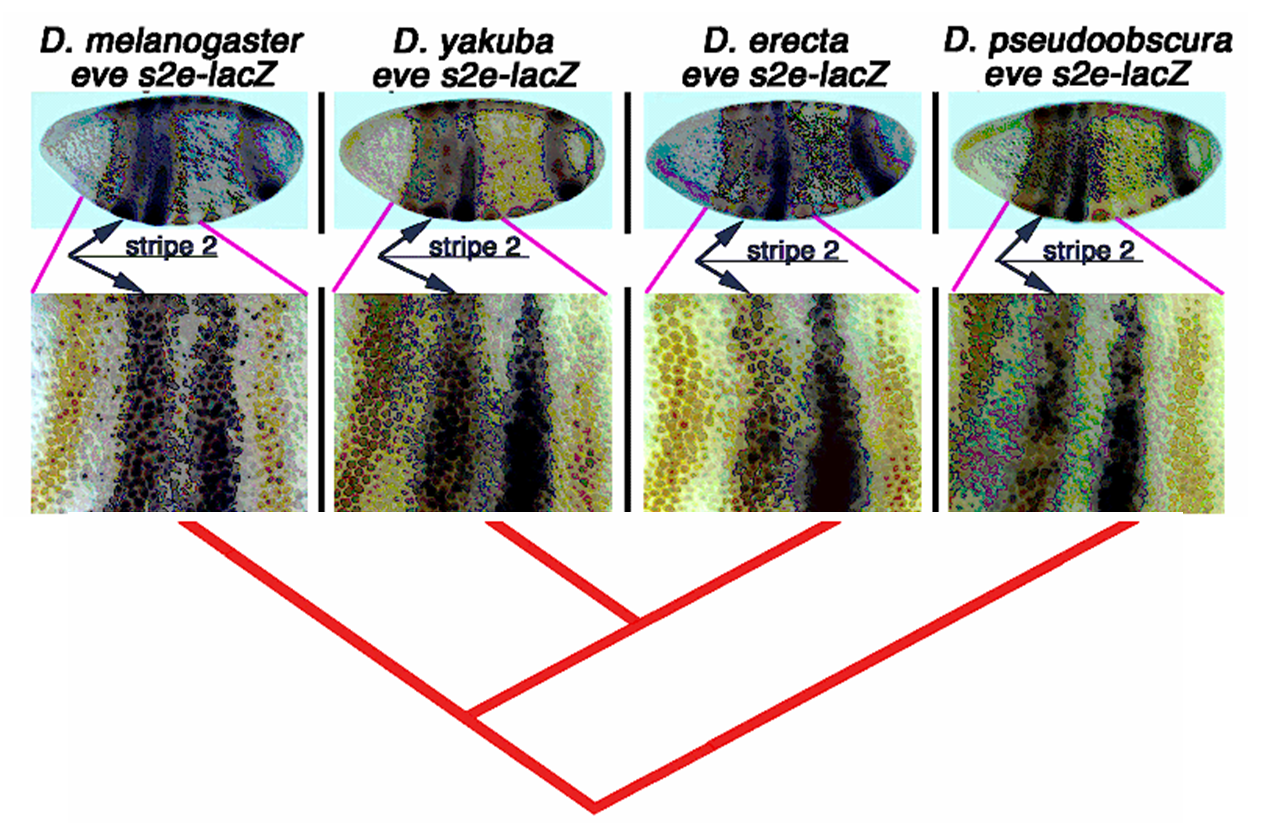 In this project we
study the evolutionary relationships
reflected in the sequence, ordering, position, spacing and function of
the regulatory motifs controlling body segmentation during early
embryogenesis in multiple species of the Drosophila. We are interested
in understanding
the biological driving forces, molecular mechanisms and functional
implications of motif evolution in general from this biological model,
and in
developing comparative genomic algorithms for motif finding from
unaligned
non-coding sequences.
In this project we
study the evolutionary relationships
reflected in the sequence, ordering, position, spacing and function of
the regulatory motifs controlling body segmentation during early
embryogenesis in multiple species of the Drosophila. We are interested
in understanding
the biological driving forces, molecular mechanisms and functional
implications of motif evolution in general from this biological model,
and in
developing comparative genomic algorithms for motif finding from
unaligned
non-coding sequences.
Selected Reading
- P.
Ray, S. Shringarpure, M. Kolar and E. P. Xing,
CSMET: Comparative Genomic Motif Detection via Multi-Resolution
Phylogenetic Shadowing ,
PLoS
Computational Biology, 4(6): e1000090.
- J-Y
Pan, A. Balan, E.P. Xing, A. Traina and C. Faloutsos, Automatic
Mining of Fruit Fly Embryo Images, The Twelfth ACM SIGKDD
International Conference on Knowledge Discovery and Data Mining (KDD
2006).
[Top]
Inference of Population Genetic Structure, Variation, Migration, and Evolution Based on Genome Polymorphisms
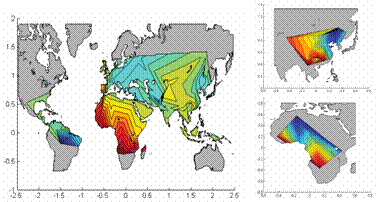 In this project we
develop novel statistical
models and computational algorithms based on formalisms such as the
coalescent
process, Bayesian nonparametrics, admixture models, and various
temporal/spatial
stochastic processes for uncovering the chromosomal association (i.e.,
haplotypes), population distribution (i.e., diversity and frequency)
inheritance
process (i.e., recombination/substitution/selection), and migration
history of
genetics polymorphisms such as SNPs, to address problems such as
ancestry
reconstruction, disease-related gene flow, chromosomal evolution and
genetic demography in human or organismal populations.
In this project we
develop novel statistical
models and computational algorithms based on formalisms such as the
coalescent
process, Bayesian nonparametrics, admixture models, and various
temporal/spatial
stochastic processes for uncovering the chromosomal association (i.e.,
haplotypes), population distribution (i.e., diversity and frequency)
inheritance
process (i.e., recombination/substitution/selection), and migration
history of
genetics polymorphisms such as SNPs, to address problems such as
ancestry
reconstruction, disease-related gene flow, chromosomal evolution and
genetic demography in human or organismal populations.
Selected Reading
- S.
Shringarpure and E. P. Xing, mStruct:
A New Admixture Model for
Inference of Population Structure in Light of Both Genetic Admixing and
Allele Mutations ,
Proceedings of the 25th International Conference on Machine Learning (ICML
2008). (A longer version
is available soon in CMU-MLD Technical
Report 08-105
with the same title.)
- K.
Sohn and E.
P. Xing, Spectrum:
Joint Bayesian
Inference of Population Structure and Recombination
Event,
The Fifteenth International Conference on Intelligence Systems for
Molecular Biology (ISMB 2007).
[Top]
Biological sequence analysis: motif detection, gene finding and systems biology
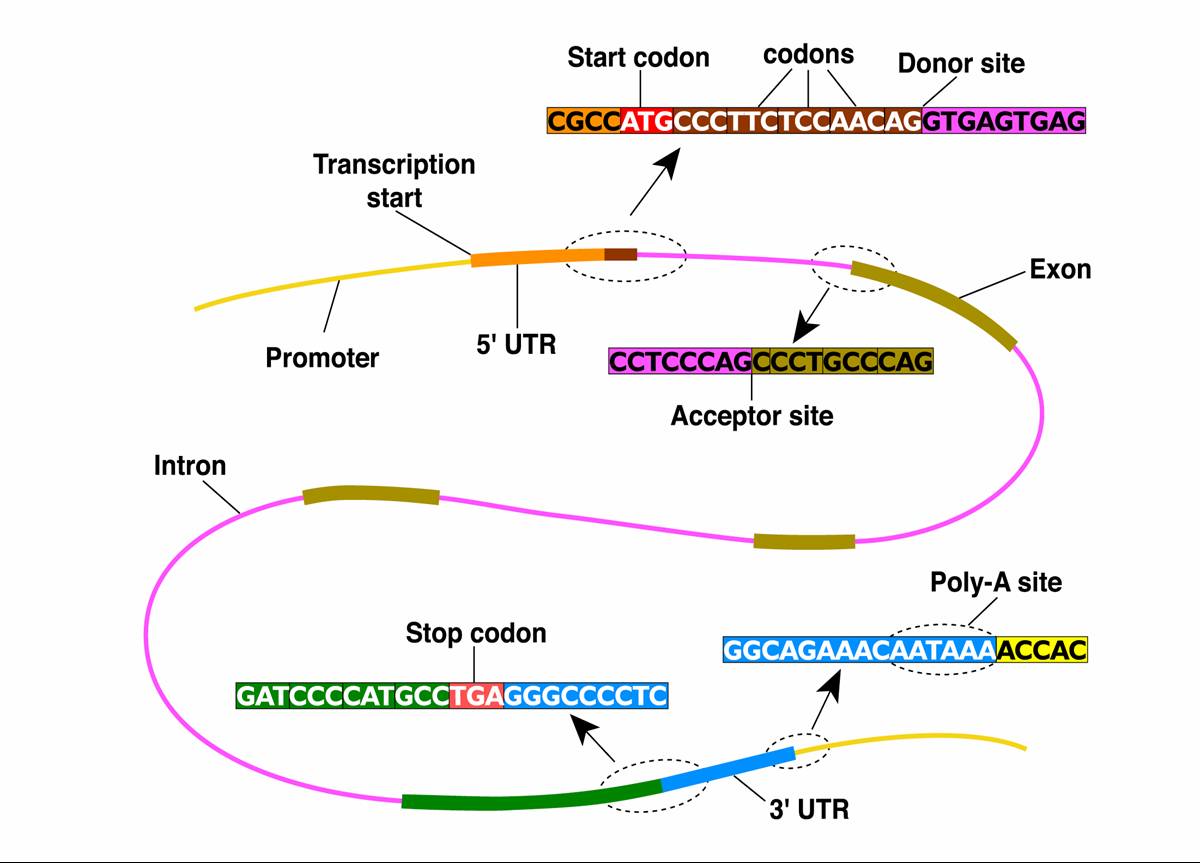 In this project we
develop models and
algorithms for
understanding and uncovering the structure of genomic sequences of
higher
organisms. We develop Bayesian models for DNA/protein motif detection
and gene
finding based on both sequence-level signatures and meta-sequence-level
structural information reflecting protein-DNA binding, transcript
stability,
and prior knowledge of the organization rules of regulatory modules. We
intend
to integrate motif finding with the system biology research of gene
regulatory
network.
In this project we
develop models and
algorithms for
understanding and uncovering the structure of genomic sequences of
higher
organisms. We develop Bayesian models for DNA/protein motif detection
and gene
finding based on both sequence-level signatures and meta-sequence-level
structural information reflecting protein-DNA binding, transcript
stability,
and prior knowledge of the organization rules of regulatory modules. We
intend
to integrate motif finding with the system biology research of gene
regulatory
network.
Selected Reading
- P.
Ray, S. Shringarpure, M. Kolar and E. P. Xing,
CSMET: Comparative Genomic Motif Detection via Multi-Resolution
Phylogenetic Shadowing ,
PLoS
Computational Biology, 4(6): e1000090.
- T.
Lin, P. Ray, G. K. Sandve, S. Uguroglu, and E. P. Xing, BayCis:
a Bayesian
hierarchical HMM
for cis-regulatory module decoding in metazoan genomes, Proceedings of the Twelfth Annual
International Conference on Research in Computational Molecular
Biology (RECOMB2008).
[Top]
|
Last updated 02/1/2009 |